Pharmacology: Pharmacodynamics: Erythropoietin is a glycoprotein that is the primary regulator of erythropoiesis. The production of erythropoietin primarily occurs in the kidney and is regulated in response to changes in tissue oxygenation. Endogenous erythropoietin production is impaired in patients with chronic renal failure (CRF) and erythropoietin deficiency is the primary cause of their anaemia.
Erythropoietin acts through specific interaction with the erythropoietin receptor on erythroid progenitor cells in the bone marrow. Using a panel of human tissues neither darbepoetin alfa nor r-HuEPO (or their desialylated forms) bound to human tissues other than those expressing the erythropoietin receptor.
NESP has been shown to stimulate erythropoiesis in anaemic CRF and cancer patients, resulting in the correction and maintenance of haemoglobin. Treatment of anaemia of CRF and cancer has been associated with a reduction in red blood cell (RBC) transfusions and improved quality of life.
In patients with cancer receiving concomitant chemotherapy, the aetiology of anaemia is multifactorial, with erythropoietin deficiency and a blunted response of erythroid progenitor cells to endogenous erythropoietin contributing significantly towards their anaemia.
Due to its increased sialic acid-containing carbohydrate content, NESP has an approximately 3-fold longer terminal half-life than erythropoietin and consequently a greater in vivo biologic activity when administered by either the subcutaneous (SC) or intravenous (IV) route.
In cancer patients with anaemia (mean ± sd haemoglobin 9.9±0.9 g/dL), a range of weekly subcutaneous doses of darbepoetin alfa from 0.5 to 8.0 μg/kg were assessed, beginning on day 1 of chemotherapy (before starting chemotherapy) and continuing for 12 weeks. Data from these studies indicate that there is a dose relationship with respect to haemoglobin response. The minimally effective starting dose with respect to reducing transfusion requirements was 1.5 μg/kg/week with a plateau observed at 4.5 μg/kg/week.
CLINICAL TRIAL: Clinical Experience in CRF Patients:
Ten clinical studies were conducted, involving SC and IV administration of darbepoetin alfa to a total of 1578 adult CRF patients with an exposure of 942 patients years. Response to darbepoetin alfa was consistent across all studies. The time to reach the target haemoglobin is a function of the baseline haemoglobin and the rate of haemoglobin rise. The rate of increase in haemoglobin is dependent upon the dose of darbepoetin alfa administered and individual patient variation.
Maintenance in CRF Patients: Darbepoetin alfa was at least equivalent to r-HuEPO in the maintenance of a target haemoglobin (haemoglobin between 9 to 13 g/dL and between -1.0 g/dL and +1.5 g/dL of baseline) in 2 trials in which adult dialysis patients were randomised to either stay on r-HuEPO or switch to darbepoetin alfa.
One trial evaluated 224 darbepoetin alfa-treated patients and 112 r-HuEPO-treated patients. The median darbepoetin alfa dose was 30 μg/week and the median r-HuEPO dose was 6000 U/week. The drugs were administered either IV or SC at frequencies varying from 3 times weekly to once every 2 weeks. Ninety-seven percent of patients in the darbepoetin alfa group received their treatment at a lower frequency than they had previously received r-HuEPO, in most cases once weekly instead of 2 to 3 times weekly. The mean difference for change in haemoglobin from baseline (darbepoetin alfa minus r-HuEPO) was 0.03 g/dL (95% confidence interval [CI]: -1.6, 2.1).
In the second trial, 121 darbepoetin alfa-treated patients and 240 r-HuEPO-treated patients were evaluated. Both drugs were administered IV, darbepoetin alfa once weekly and r-HuEPO 3 times weekly. The median darbepoetin alfa dose was 38 μg/week and the median r-HuEPO dose 9900 U/week. The mean difference for change in haemoglobin from baseline (darbepoetin alfa minus r-HuEPO) was 0.16 g/dL (95% CI: -0.8, 3.3).
There were no significant differences between the drugs in the proportion of patients with unstable haemoglobin and proportion receiving blood transfusions, in either trial.
Correction of Anaemia in CRF Patients:
In a trial in adult predialysis CRF patients with anaemia (haemoglobin concentration <11 g/dL), darbepoetin alfa produced a similar response to r-HuEPO with 87% (95% CI: 80, 92) of darbepoetin alfa-treated patients (n=129) and 86% (95% CI: 71, 95) of r-HuEPO treated patients (n=37) achieving the haemoglobin target (>11 g/dL and >1 g/dL increase from baseline) after 16 weeks. The drugs were administered by the SC route. The starting dose of darbepoetin alfa was 0.45 μg/kg once weekly (approximately equivalent to 90 U/kg of r-HuEPO weekly). The starting dose of r-HuEPO was 50 U/kg twice weekly (100 U/kg total weekly dose). The doses were adjusted in ±25% increments at 2 to 4 week intervals as required. The median time to response was 7 weeks in each group and the median doses at response were similar to the starting doses, 0.46 μg/kg/week for darbepoetin alfa and 100 U/kg/week for r-HuEPO. The median dose after 16 weeks of treatment was 0.45 μg/kg/week for darbepoetin alfa and 100 U/kg/week for r-HuEPO.
In a second trial, in adult dialysis CRF patients with anaemia (haemoglobin <10 g/dL), r-HuEPO was started at a higher dose than darbepoetin alfa based on protein mass, 50 U/kg 3 times weekly (150 U/kg total weekly dose) compared with 0.45 μg/kg once weekly (approximately equivalent to 90 U/kg of r-HuEPO weekly). The drugs were administered either IV or SC. A similar regime of dosage adjustments and a similar haemoglobin target were employed to the previous trial. Of patients receiving at least one dose of drug, 95% (95% CI: 77, 100) of r-HuEPO-treated patients (n=22) and 71% (95% CI: 59, 82) of darbepoetin alfa-treated patients (n=70) reached the haemoglobin target by 20 weeks. The median time to response was 8 weeks in the r-HuEPO group and 9 weeks in the darbepoetin alfa group and the median doses at response were 150 U/kg/week and 0.55 μg/kg/week, respectively. The median dose after 20 weeks of treatment was 0.56 μg/kg/week for darbepoetin alfa and 150 U/kg/week for r-HuEPO.
Treatment of Anaemia in Cancer Patients Receiving Chemotherapy: A randomised, double-blind, placebo-controlled, parallel-group trial was conducted in anaemic patients with lung cancer receiving multi-cycle platinum-containing chemotherapy. Randomisation was stratified by tumour type (small cell, non-small cell) and region (Australia, Canada, Central and Eastern Europe, Western Europe). The starting dose was 2.25 μg/kg/week as a single subcutaneous injection commencing on day 1 prior to administration of chemotherapy. The dose could be increased after 6 weeks up to 4.5 μg/kg/week if patients failed to achieve an increase in haemoglobin of >1 g/dL. The duration of treatment was 12 weeks.
Efficacy was determined by a reduction in the proportion of patients who were transfused over the 12-week treatment period. A significantly lower proportion of patients in the darbepoetin alfa arm, 26% (95% CI: 20, 33) required transfusion compared to 60% (95% CI: 52, 68) in the placebo arm (Kaplan-Meier estimate of proportion; p <0.001 by Cochran-Mantel-Haenszel test) (see Table 2). There was a trend in favour of darbepoetin alfa in FACT/F, a fatigue-related quality of life score. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
There were 67 patients in the darbepoetin alfa arm who had their dose increased from 2.25 to 4.5 μg/kg/week, at any time during the treatment period. Of the 67 patients who received a dose increase, 28% had a 2 g/dL increase in haemoglobin over baseline, generally occurring between weeks 8 to 13. Of the 89 patients who did not receive a dose increase, 69% had a 2 g/dL increase in haemoglobin over baseline, generally occurring between weeks 6 to 13.
In the same study, the effect of darbepoetin alfa on tumour progression and survival was evaluated through long-term surveillance of patients. After a median observation period of approximately 1 year, the median time to disease progression in the darbepoetin alfa group (n=155) was 29 weeks (95% CI: 22, 33) compared with 22 weeks (95% CI: 18, 25) in the placebo group (n=159). The median time to death in the darbepoetin alfa group was 43 weeks (95% CI: 37, not estimable) compared with 35 weeks (95% CI: 29, 48) in the placebo group.
Geriatric Use: More than 1500 darbepoetin alfa-treated patients with CRF have been studied; 28% were 65 to 74 years of age and 15% were 75 years or older. Of the 781 cancer patients in clinical studies receiving darbepoetin alfa and concomitant chemotherapy, 31% were age 65 to 74 years of age, while 12% were 75 and over. No differences in dose requirements, safety or efficacy were observed between geriatric and younger adult patients.
Multinational Clinical Study in Myelodysplastic Syndrome: NESP was subcutaneously administered to 52 patients with myelodysplastic syndromes who were in the low or intermediate-1 risk categories under IPSS and transfusion-dependent
note 1) with the serum erythropoietin concentration of 500 mIU (international units)/mL or lower at a dose of 60, 120, or 240μg once weekly for 48 weeks
note 2). The efficacy of NESP was assessed at 16 weeks after the initiation of NESP administration
note 3). In the 50 patients included in efficacy evaluation, major erythroid response
note 4) or minor erythroid response
note 5) was observed in 11 of 17 patients (64.7%) of the 60μg group, 8 of 18 patients (44.4%) of the 120μg group, and 10 of 15 patients (66.7%) of the 240μg group.
Note 1) Defined as the longest transfusion-free interval of shorter than 56 days in the past 112 days (excluding transfusions performed when the haemoglobin concentration was higher than 9.0 g/dL).
Note 2) If patients did not respond to NESP at 16 weeks after the initiation of administration, administration of NESP was discontinued in the 240 μg group, and the dose was increased in the other groups.
Note 3) The target haemoglobin concentration was set at 10.0 g/dL by reference to the Guidelines for use of blood products, revised version (in Japanese) (Blood and Blood Products Division, PFSB, MHLW, 2005). To maintain the haemoglobin within the target range of 9.0 to 11.0 g/dL, administration of NESP was suspended if the haemoglobin concentration exceeded 11.0 g/dL.
Note 4) Defined as transfusion independence for at least 56 consecutive days during the NESP administration period, and the maximum haemoglobin concentration during the transfusion-free period of at least 1.0 g/dL higher than that at the initiation of administration.
Note 5) Defined as 50% decrease or more in transfusion requirement in 56 consecutive days during the NESP administration period in comparison with during the 56-day period before the initiation of administration. (See Tables 3, 4, 5, 6 and 7.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacokinetics: General: The concentration of darbepoetin alfa in the circulation remains above the minimum stimulatory concentration for erythropoiesis for longer than an equivalent molar dose of r-HuEPO. This allows darbepoetin alfa to be administered less frequently to achieve the same biological response. The pharmacokinetic properties of darbepoetin alfa have been studied in healthy adult subjects, in adult and paediatric CRF patients, and in adult cancer patients. In all cases darbepoetin alfa exhibits dose-linearity over the therapeutic dose range.
Subcutaneous absorption: Following SC administration in adult CRF patients, the absorption is slow and rate-limiting. The peak concentration occurs at 34 hours (range: 24 to 72 hours) post-SC administration in adult CRF patients, and bioavailability is approximately 37% (range: 30% to 50%). After SC administration of 2.25 μg/kg to adult cancer patients, darbepoetin alfa reached peak concentration at a median of 94.5 hours (range: 70.8 to 123 hours).
Distribution following intravenous administration: Distribution of darbepoetin alfa in adult CRF patients is predominantly confined to the vascular space (approximately 60 mL/kg). The distribution half-life, following IV administration, is 1.4 hours.
Elimination: In adult CRF patients, the terminal half-life of darbepoetin alfa following IV administration is approximately 21 hours (range: 12 to 40 hours). Following SC administration, the terminal half-life is 49 hours (range: 27 to 89 hours) and 39.2 hours (range: 15.5 to 54.2) in CRF and cancer patients, respectively, reflecting the long absorption half-life.
Multiple dosing: With once weekly dosing in adult CRF patients, steady-state serum concentrations are achieved within 4 weeks with <2-fold increase in peak concentration. Accumulation was negligible following both SC and IV dosing over 1 year of treatment.
In adult cancer patients, the pharmacokinetic properties did not change with multiple dosing over 12 weeks (dosing every week or every 2 weeks). The expected moderate increases (less than 2-fold) in darbepoetin alfa serum concentrations upon multiple dosing were observed as steady state was approached. No unexpected accumulation was observed upon repeated administration of darbepoetin alfa across a wide range of doses at once weekly and once every 2 weeks dosing schedules.
Special Populations: Paediatric:
The pharmacokinetic parameters of darbepoetin alfa in paediatric CRF patients are similar to adult CRF patients. Following SC or IV administration in children 7 to 16 years old, the terminal half-life was 21 hours (range: 12 to 25 hours) for IV administration and 33 hours (range: 16 to 44 hours) for SC administration. The SC bioavailability was 52% (range: 32% to 70%).
Hepatic dysfunction: The efficacy and safety of darbepoetin alfa have not been established in patients with hepatic dysfunction.
Myelodysplastic Syndrome: Single administration (Japanese and Korean patients): Following repeated subcutaneous administration of NESP at doses of 60-240 μg to patients with myelodysplastic syndrome for 16 weeks, the time course of serum concentrations and pharmacokinetic parameters at the initial administration were as follows. C
max and AUC
0-t did not increase in proportion to the dose. (See figure and Table 8.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Repeated administration (Japanese and Korean patients): Following repeated subcutaneous administration of NESP at doses of 60 to 240 μg to patients with myelodysplastic syndromes for 16 weeks, the serum trough concentration was not dose proportional and showed no remarkable changes over the dose range tested throughout the administration period.
Toxicology: Preclinical Experience: Darbepoetin alfa undergoes extensive metabolism, with less than 2% of intact darbepoetin alfa being excreted renally in rats, while degradation products are recovered in the urine (57% dose) and faeces (24% dose). Metabolism of darbepoetin alfa may involve desialylation by blood/tissue sialidases, with subsequent rapid removal of the desialylated form by hepatic receptors, and/or reuptake via bone marrow cells.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image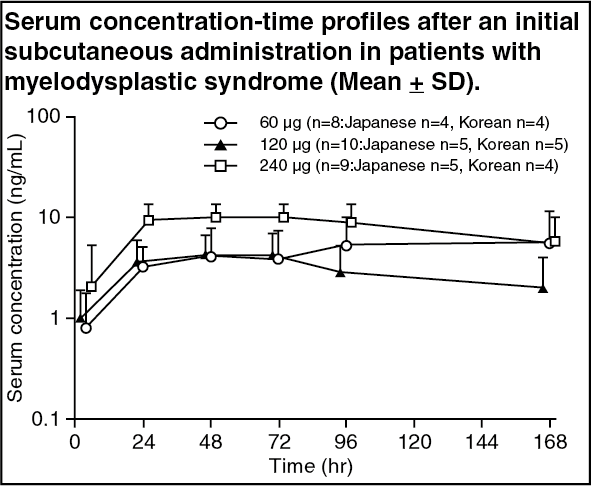 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image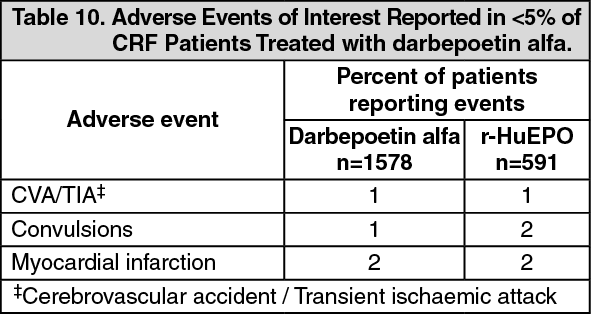 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image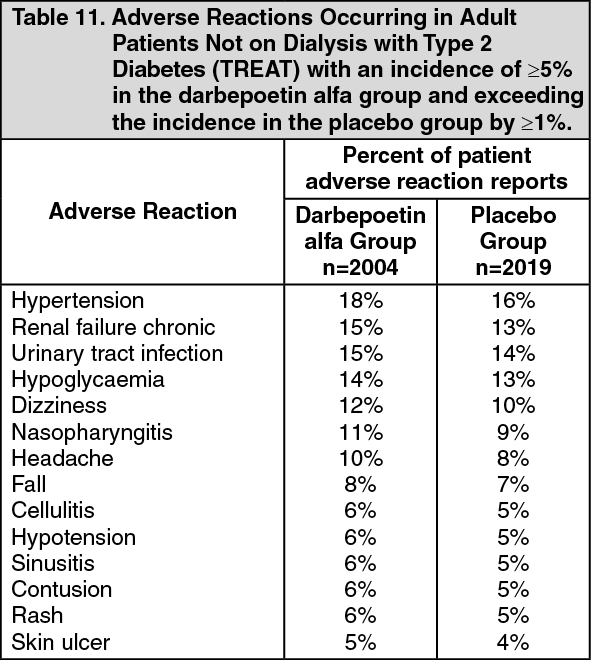 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image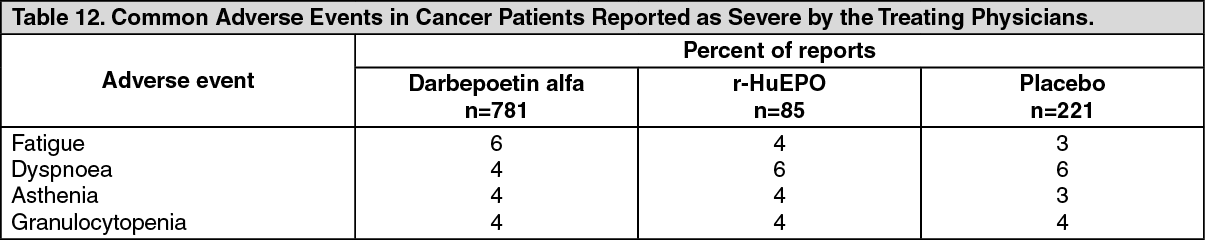 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out
