ACTIVE INGREDIENT: Ferric Derisomaltose 417 mg/ml eqv Elementary Iron 100 mg/ml.
Ferric derisomaltose is a dark reddish brown powder containing 24% iron (III). Ferric derisomaltose solution for injection is a colloid with iron in spheroidal iron-carbohydrate particles. The structure of the ferric derisomaltose particle has been characterised by carbon 13 NMR spectroscopic analysis which reveals that the complex forms a stable matrix type structure with about 10 iron(III) atoms to one molecule of isomaltoside pentamer with the iron(III) bound in cavities of the 3-D structure of isomaltoside pentamers, leading to a low content of free iron based on in vitro data.
Chemical name: Iron (III) hydroxide isomaltoside 1000.
Synonyms: iron isomaltoside, iron isomaltooligosaccharide, iron oligosaccharide, iron isomaltopentaoside.
Molecular Formula: {FeO(1-3X)(OH)(1+3X)(C6H5O73-)X}, (H20)T, -(C6H10O6)R(-C6H10O5-)Z(C6H13O5)R, (NaCl)Y X= 0.0311; T = 0.25; R = 0.14; Z = 0.49; Y = 0.14.
This unit has a molecular weight of 235 g/mole - The molecular weight of the colloid particle is approximately 165 kDa hence the colloid particle contains approximately 700 repeatable units (165,000/235).
CAS number: 1345510-43-1.
Monofer is a sterile, dark brown, non-transparent solution with pH 5.0-7.0 containing ferric derisomaltose dissolved in water for injection. The pH may have been adjusted with hydrochloric acid or sodium hydroxide, and the product is filled under nitrogen gas.
Excipients/Inactive Ingredients: Water for injections, Sodium hydroxide, Hydrochloric acid.
Pharmacotherapeutic group: Iron parenteral preparation. ATC code: B03AC.
Pharmacology: Pharmacodynamics: The isomaltoside 1000 component of ferric derisomaltose consists of 3-5 glucose units with an average molecular weight of approximately 1000 Da. It has no detectable branching structures as evidenced by careful 13C and 1H NMR spectroscopic analysis. Furthermore isomaltoside 1000 does not contain any reducing sugar residues, which can be involved in complex redox reactions.
Linear carbohydrates with a molecular weight of less than 1300 have been shown to not induce immune responses in vivo.
The Monofer formulation contains iron in a complex with isomaltoside 1000 that releases bioavailable iron to iron-binding proteins.
The iron is available in a non-ionic water-soluble form in an aqueous solution with pH between 5.0 and 7.0.
Evidence of a therapeutic response can be seen within a few days of administration of ferric derisomaltose as an increase in the reticulocyte count. Due to the release of bioavailable iron serum ferritin peaks within days after an intravenous dose of Monofer and slowly returns to baseline after weeks.
Clinical trials: The efficacy of Monofer has been studied in the different therapeutic areas necessitating intravenous iron to correct iron deficiency. The main trials are described in more detail as follows.
Iron deficiency anaemia outside CKD: The P-Monofer-IDA-01 trial was an open-label, comparative, randomised, multi-centre, non-inferiority trial conducted in 511 patients with IDA randomised 2:1 to either Monofer or iron sucrose. 90% of recruited patients were females. The dosing of Monofer was performed according to the Simplified Table as described under Dosage & Administration and dosing of iron sucrose was calculated according to Ganzoni and administered as 200 mg infusions. The primary endpoint was the proportion of patients with an Hb increase ≥2.0 g/L from baseline at any time between weeks 1 to 5. A higher proportion of patients treated with Monofer compared to iron sucrose reached the primary endpoint, 68.5% vs 51.6%, respectively (Full Analysis Set (FAS), p < 0.0001).
Nephrology: Non-dialysis-dependent chronic kidney disease: The P-Monofer-CKD-02 trial was an open-label, comparative, randomised, multi-centre, non-inferiority trial conducted in 351 iron deficient non-dialysis dependent (NDD) chronic kidney disease (CKD) patients, randomised 2:1 to either Monofer or oral iron sulphate administered as 100 mg elemental oral iron twice daily (200 mg daily) for 8 weeks. The patients in the Monofer group were randomised to infusion of 1000 mg single doses or bolus injections of 500 mg. The test for non-inferiority showed that Monofer was non-inferior to iron sulphate in its ability to increase Hb from baseline to week 4 in both FAS and PP datasets (FAS: difference estimate: 0.2216, 95% CI: 0.012:0.431, p < 0.0001; PP: difference estimate: 0.2176, 95% CI: 0.003:0.432, p < 0.0001) and also sustained a superior increase in Hb compared to oral iron from week 3 until the end of trial at week 8 (p=0.009 at week 3, p=0.04 at week 4, p=0.0004 at week 8, FAS).
Haemodialysis-dependent chronic kidney disease: The P-Monofer-CKD-03 trial was an open-label, comparative, randomised, multi-centre, non-inferiority trial conducted in 351 haemodialysis patients randomised 2:1 to either Monofer or iron sucrose. Patients were randomised to either a single injection of 500 mg or 500 mg in split doses of Monofer or 500 mg iron sucrose in split doses. In the FAS of 341 subjects, a total of 187 (82.7%) subjects treated with ferric derisomaltose and 95 (82.6%) subjects treated with iron sucrose were able to maintain Hb between 95 and 125 g/L.
Oncology: Cancer related anaemia: The P-Monofer-CIA-01 trial was an open-label, comparative, randomised, multi-centre, non-inferiority trial conducted in 350 cancer patients with anaemia randomised 2:1 to either Monofer or oral iron sulphate administered as 100 mg elemental oral iron twice daily (200 mg daily) for 12 weeks. The patients in the Monofer group were randomised to either an infusion of max 1000 mg single doses or bolus injections of 500 mg. The primary endpoint was change in Hb concentrations from baseline to week 4. The test for non-inferiority showed that Monofer was non-inferior to iron sulphate in its ability to increase Hb from baseline to week 4 in both FAS and PP datasets (FAS: difference estimate: 0.0161, 95% CI: -0.261:0.293, p = 0.0002; PP: difference estimate: -0.0071, 95% CI: -0.291:0.276, p = 0.0006).
Gastroenterology: Inflammatory bowel disease: The P-Monofer-IBD-01 trial was an open-label, comparative, randomised, multi-centre, non-inferiority trial conducted in 338 inflammatory bowel disease (IBD) patients randomised 2:1 to receive either Monofer or oral iron sulphate administered as 100 mg elemental oral iron twice daily for 8 weeks (200 mg daily). The patients in the Monofer group were randomised to either an infusion of max 1000 mg single doses or bolus injections of 500 mg. The primary endpoint was change in Hb concentrations from baseline to week 8. A modified Ganzoni formula was used to calculate the IV iron need with a target Hb of only 130 g/L. Non-inferiority could not be statistically demonstrated on the primary endpoint (FAS: p = 0.0945). The study demonstrated an increase in Hb concentration from a mean of 96.4 g/L at baseline to 122.3 g/L at week 8 in subjects treated with Monofer and an increase from 96.1 g/L at baseline to 125.9 g/L at week 8 in subjects treated with oral iron sulphate. The dose-response relationship observed with Monofer suggests that the true iron demand of IV iron was underestimated by the modified Ganzoni formula.
Post-partum haemorrhage: The P-Monofer-PP-01 trial was an open-label, comparative, randomised, single-centre trial conducted in 200 healthy women with postpartum haemorrhage exceeding 700 mL within 48 hours after delivery. The women were randomised 1:1 to receive either a single dose of 1200 mg Monofer or standard medical care (i.e the majority of patients were prescribed oral iron). The primary endpoint was the aggregated change in physical fatigue within 12 weeks postpartum. The difference in aggregated change in physical fatigue score within 12 weeks postpartum was -0.97 (p=0.006), in favour of Monofer, although the estimated treatment difference was less than the pre-defined minimum clinically relevant difference of 1.8 required for claiming superiority.
Surgery: Non-anaemic patients undergoing cardiac surgery: The P-Monofer-CABG-01 trial was a double-blind, placebo-controlled, randomised, single-centre trial of 60 non-anaemic patients undergoing cardiac surgery (coronary artery bypass graft, valve replacement, or a combination thereof). The patients were randomized 1:1 to either 1000 mg Monofer administered perioperatively by infusion or placebo. The primary endpoint was to assess the change in Hb concentrations from baseline to 4 weeks postoperatively. One month after surgery, the decrease in haemoglobin concentration was less pronounced in the Monofer treated group, with an average of 126 g/L versus 118 g/L (p=0.012) and significantly more patients were non-anaemic in the Monofer treated group compared to the placebo group (38.5% versus 8.0%; p=0.019).
Pharmacokinetics: There is no data investigating the pharmacokinetics of single and multiple doses of MONOFER above 1000 mg.
Ferric derisomaltose displays inter-patient variability in PK parameters, including AUC and Tmax.
Distribution: Following intravenous administration, ferric derisomaltose or released free iron is taken up by the cells of the reticuloendothelial system (RES), particularly in the liver and spleen, from where iron is slowly released.
Metabolism: Circulating iron is removed from the plasma by cells of the RES. The iron binds available protein moieties to form hemosiderin or ferritin, the physiological storage forms of iron, or to a lesser extent, the transport molecule transferrin. This iron, which is subject to physiological control, replenishes haemoglobin (Hb) and depleted iron stores.
Excretion: After administration of a single dose of ferric derisomaltose of 100 to 1000 mg of iron in the pharmacokinetic studies, the iron injected or infused was cleared from the plasma with a half-life that ranged from 1 to 4 days. Renal elimination of iron was negligible.
Iron is not easily eliminated from the body and accumulation can be toxic. Due to the size of the complex, ferric derisomaltose is not eliminated via the kidneys. Small quantities of iron are eliminated in urine and faeces.
Isomaltoside 1000 is either metabolised or excreted.
Toxicology: Preclinical safety data: Genotoxicity: Ferric derisomaltose was not genotoxic in a bacterial mutation assay, a chromosomal aberration test in human lymphocytes in vitro or a micronucleus assay in mice in vivo.
Carcinogenicity: Carcinogenicity studies were not conducted.
Monofer is indicated for the treatment of iron deficiency in adults, under the following conditions: When oral iron preparations are ineffective or cannot be used.
Where there is a clinical need to deliver iron rapidly.
The diagnosis must be based on laboratory tests.
Dosage: Calculation of the cumulative iron need:
Iron replacement in patients with iron deficiency: The dose of Monofer is expressed in mg of elemental iron. The iron need and the administration schedule for Monofer must be individually established for each patient. The optimal Hb target level and iron stores may vary in different patient groups and between patients. Refer to official guidelines.
Iron deficiency anaemia will not appear until essentially all iron stores have been depleted. Iron therapy should therefore replenish both Hb iron and iron stores.
After the current iron deficit has been corrected, patients may require continued therapy with Monofer to maintain target levels of Hb and acceptable limits of other iron parameters.
The cumulative iron need can be determined using either the Ganzoni formula (1) or Table 1 as follows (2). It is recommended to use the Ganzoni formula in patients who are likely to require individually adjusted dosing such as patients with anorexia nervosa, cachexia, obesity, pregnancy or anaemia due to bleeding.
1. Ganzoni formula: (See equation.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The dose-response relationship observed with Monofer suggests that the true iron demand of IV iron is underestimated by the Ganzoni fomula if a Hb target of less than 150 g/L is used.
2. Simplified Table: (See Table 1.)
Click on icon to see table/diagram/image
Method of administration: Monofer must only be mixed with sterile 0.9% sodium chloride. No other intravenous dilution solutions should be used. No other therapeutic agents should be added. The diluted solution for injection should be visually inspected prior to use. Use only clear solutions without sediment.
Monitor carefully patients for signs and symptoms of hypersensitivity reactions during and following each administration of Monofer.
Each IV iron administration is associated with a risk of a hypersensitivity reaction. Thus, to minimise risk the number of single IV iron administrations should be kept to a minimum.
Monofer offers the flexibility of administration either as an intravenous bolus injection, as an intravenous drip infusion or as a direct injection into the venous limb of the dialyser.
Monofer should not be administered concomitantly with oral iron preparations, since the absorption of oral iron might be decreased.
Intravenous bolus injection: Monofer may be administered as an intravenous bolus injection up to 500 mg up to three times a week at an administration rate of up to 250 mg iron/minute. It may be administered undiluted or diluted in maximum 20 mL sterile 0.9% sodium chloride.
Intravenous drip infusion: The cumulative iron dose required may be administered in a single Monofer infusion up to 20 mg iron/kg body weight or as weekly infusions until the cumulative iron dose has been administered.
If the cumulative iron dose exceeds 20 mg iron/kg body weight, the dose must be split in two administrations with an interval of at least one week. It is recommended whenever possible to give 20 mg iron/kg body weight in the first administration. Dependent on clinical judgement the second administration could await follow-up laboratory tests.
Doses up to 1000 mg must be administered over 20 minutes. Doses exceeding 1000 mg must be administered over 30 minutes or more. Single doses above 1500 mg are not recommended.
Monofer should be added to maximum 500 mL sterile 0.9% sodium chloride.
Injection into dialyser: Monofer may be administered during a haemodialysis session directly into the venous limb of the dialyser under the same procedures as outlined for intravenous bolus injection.
The ferric derisomaltose in Monofer has a low toxicity. The preparation is well tolerated and has a minimal risk of accidental overdosing.
Overdose may lead to accumulation of iron in storage sites eventually leading to haemosiderosis. Monitoring of iron parameters such as serum ferritin may assist in recognising iron accumulation. Supportive measures such as chelating agents can be used.
Hypersensitivity to the active substance, to Monofer or any of the excipients.
Known serious hypersensitivity to other parental iron products.
Non-iron deficiency anaemia (eg. haemolytic anaemia).
Iron overload or disturbances in utilisation of iron (eg. haemochromatosis, haemosiderosis).
Decompensated liver cirrhosis or active hepatitis.
Parenterally administered iron preparations can cause hypersensitivity reactions including serious and potentially fatal anaphylactic/anaphylactoid reactions. Hypersensitivity reactions have also been reported after previously uneventful doses of parenteral iron complexes. There have been reports of hypersensitivity reactions to some parenteral iron complexes which progressed to Kounis syndrome (acute allergic coronary arteriospasm that can result in myocardial infarction).
The risk is enhanced for patients with multi allergies including drug allergies, including patients with a history of severe asthma, eczema or other atopic allergy.
There is also an increased risk of hypersensitivity reactions to parenteral iron complexes in patients with immune or inflammatory conditions (e.g. systemic lupus erythematosus, rheumatoid arthritis).
Monofer should only be administered in an environment where full resuscitation facilities can be assured. Each patient should be observed for adverse effects for at least 30 minutes following each ferric derisomaltose injection. If hypersensitivity reactions or signs of intolerance occur during administration, the treatment must be stopped immediately.
In patients with compensated liver dysfunction, parenteral iron should only be administered after careful benefit/risk assessment. Parenteral iron administration should be avoided in patients with hepatic dysfunction (alanine aminotransferase and/or aspartate aminotransferase > 3 times upper limit of normal) where iron overload is a precipitating factor, in particular Porphyria Cutanea Tarda (PCT). Careful monitoring of iron status is recommended to avoid iron overload.
Parenteral iron must be used with caution in case of acute or chronic infection, asthma, eczema or atopic allergies. It is recommended that the administration of Monofer is stopped in patients with ongoing bacteraemia. In patients with chronic infection a risk/benefit evaluation has to be performed, taking into account the suppression of erythropoiesis.
Hypotensive episodes may occur if intravenous injection is administered too rapidly.
Caution should be exercised to avoid paravenous leakage when administrating ferric derisomaltose. Paravenous leakage of ferric derisomaltose at the injection site may lead to irritation of the skin and potentially long lasting brown discoloration at the site of injection. In case of paravenous leakage, the administration of ferric derisomaltose must be stopped immediately.
Effects on laboratory tests: Parenteral iron may cause falsely elevated values of serum bilirubin and falsely decreased values of serum calcium.
Effects on ability to drive and use machines: The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
Use in Children: Ferric derisomaltose is not recommended for use in children and adolescents < 18 years due to insufficient data on safety and efficacy.
Use in the Elderly: No specific adjustment is required for use in the elderly.
A careful risk benefit assessment is required before parenteral iron is used in patients aged > 65 years and close monitoring for adverse events is required.
Effects on fertility: There are no data on the effect of ferric derisomaltose on human fertility.
In a fertility study with Ferric derisomaltose in rats no effects on female fertility or male reproductive performance and spermatogenic parameters were found at the dose levels tested.
Ferric derisomaltose did not affect fertility in male or female rats when administered IV at up to 19 mg/kg/day in males and 32 mg/kg/day in females (4 and 7 times the maximum recommended weekly clinical dose, respectively). Atrophy of prostate and seminal vesicles, seminiferous epithelium degeneration of testes, and degenerative germ cells in epididymides were observed in rats at 80 mg/kg/day thrice weekly (8 times the maximum recommended weekly clinical dose).
Use in pregnancy: There are no adequate and well-controlled trials of ferric derisomaltose in pregnant women. A careful risk/benefit evaluation is therefore required before use during pregnancy and ferric derisomaltose should not be used during pregnancy unless clearly necessary.
Iron deficiency anaemia occurring in the first trimester of pregnancy can in many cases be treated with oral iron. Treatment with ferric derisomaltose should be confined to the second and third trimester if the benefit is judged to outweigh the potential risk for both the mother and the foetus. In rare cases, foetal bradycardia has been observed in pregnant women with hypersensitivity reactions.
Iron complexes have been reported to be teratogenic and embryocidal in non-anaemic pregnant animals at high single doses above 125 mg iron/kg body weight. The highest recommended dose in clinical use is 20 mg iron/kg body weight.
In embryofetal development studies in rats and rabbits, there was a dose-dependent increase in bent ribs in rats at all doses (3-32 mg/kg/day, 0.7-8 times the maximum recommended weekly clinical dose) and fetal malformations (including hydrocephaly, microglossia, narrow pectoral region) in rabbits at >= 25 mg/kg/day (>= 6 times the maximum recommended weekly clinical dose). There was a significant reduction in viable fetuses and mean fetal weight and an increase in late resorption in rabbits at 43 mg/kg/day (10 times the maximum recommended weekly clinical dose).
Use in lactation: No formal clinical studies investigating excretion of ferric derisomaltose have been performed. In clinical study P-PP-01, a maternal milk sample was collected on day 3 from a total of 65 subjects. Overall, mean maternal milk iron level was higher in the iron isomaltoside 1000 group (0.000721 g/L) than in the standard medical care group (0.000400 g/L) at day 3. However, at week 1 the mean maternal milk iron level in the iron isomaltoside 1000 group had decreased to the same level as in the standard medical care group (0.000468 g/L and 0.000442 g/L, respectively).
Acute severe hypersensitivity reactions may occur with parenteral iron preparations. They usually occur within the first few minutes of administration and are generally characterised by the sudden onset of respiratory difficulty and/or cardiovascular collapse; fatalities have been reported. Other less severe manifestations of immediate hypersensitivity, such as urticaria and itching may also occur. In pregnancy, associated foetal bradycardia may occur with parenteral iron preparations.
Flushing in the face, acute chest and/or back pain and tightness sometimes with dyspnea in association with intravenous iron treatment may occur (frequency uncommon). This may mimic the early symptoms of an anaphylactoid/anaphylactic reaction. The infusion should be stopped and the patient's vital signs should be assessed. These symptoms disappear shortly after the iron administration is stopped. They typically do not reoccur if the administration is restarted at a lower infusion rate.
The table presents the adverse drug reactions (ADRs) reported during ferric derisomaltose treatment in clinical trials and in-market experience. (See Table 2.)
Click on icon to see table/diagram/image
Description of selected adverse reactions: Delayed reactions may also occur with parenteral iron preparations and can be severe. They are characterised by arthralgia, myalgia and sometimes fever. The onset varies from several hours up to four days after administration. Symptoms usually last two to four days and settle spontaneously or following the use of simple analgesics.
Other Post-Marketing experience: Because these adverse events are spontaneously reported in a voluntary manner from a population of uncertain size, it is not possible to reliably estimate their frequency.
Cardiac disorders: Cardiac arrest.
General disorders and administration site conditions: Asthenia, chest discomfort, feeling abnormal, feeling hot.
Musculoskeletal and connective tissue disorders: Joint swelling, pain in extremity.
Nervous system disorders: Burning sensation, cerebrovascular accident, generalized tonic-clonic seizure, syncope.
Respiratory, thoracic and mediastinal disorders: Asphyxia, pharyngeal edema, respiratory arrest, respiratory distress, wheezing.
Skin and subcutaneous tissue disorders: Erythema, generalized erythema, purpura, skin discolouration, swelling face.
Vascular disorders: Shock.
Post-marketing experience: Post marketing adverse reactions are included in the previous table.
Reporting suspected adverse effects: Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product.
As with all parenteral iron preparations the absorption of oral iron is reduced when administered concomitantly. Oral iron therapy should not be started earlier than 5 days after the last injection of ferric derisomaltose.
Large doses of parenteral iron (5 mL or more) have been reported to give a brown colour to serum from a blood sample drawn four hours after administration.
Incompatibilities: Monofer must only be mixed with sterile 0.9% sodium chloride. No other intravenous dilution solutions should be used. No other therapeutic agents should be added.
Special precautions for disposal: Monofer is for single use in one patient only. Discard any residue.
Shelf life: Shelf life after dilution with sterile 0.9% sodium chloride: Diluted solutions should be used immediately.
Store below 30°C.
B03AC - Iron, parenteral preparations ; Used in the treatment of anemia
MonoFer soln for inj/infusion 100 mg/mL
5 mL x 5 × 1's


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image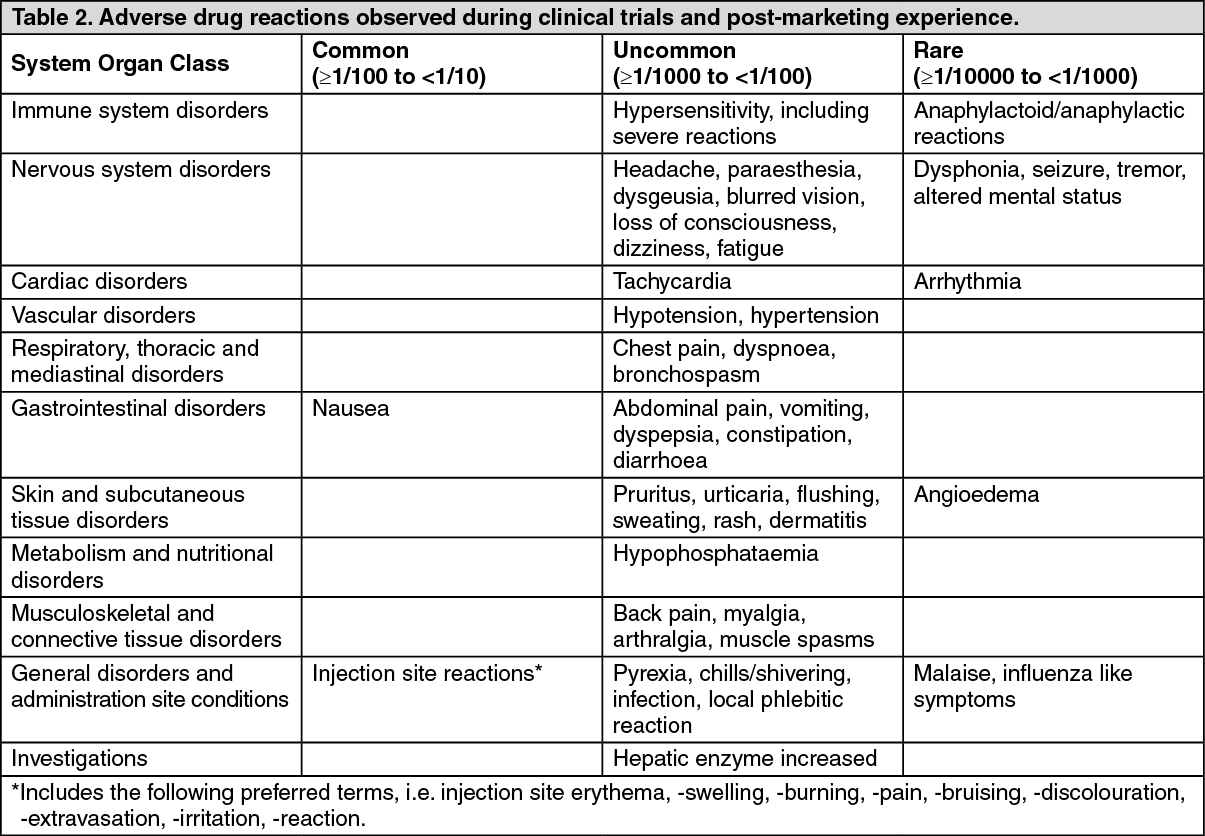 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
