


 Sign Out
Sign Out



Pharmacology: Pharmacodynamics: Rosuvastatin: Mechanism of action: Rosuvastatin is a selective and competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts 3-hydroxy-3-methylglutaryl coenzyme A to mevalonate, a precursor for cholesterol. The primary site of action of rosuvastatin is the liver, the target organ for cholesterol lowering.
Rosuvastatin increases the number of hepatic LDL receptors on the cell-surface, enhancing uptake and catabolism of LDL and it inhibits the hepatic synthesis of VLDL, thereby reducing the total number of VLDL and LDL particles.
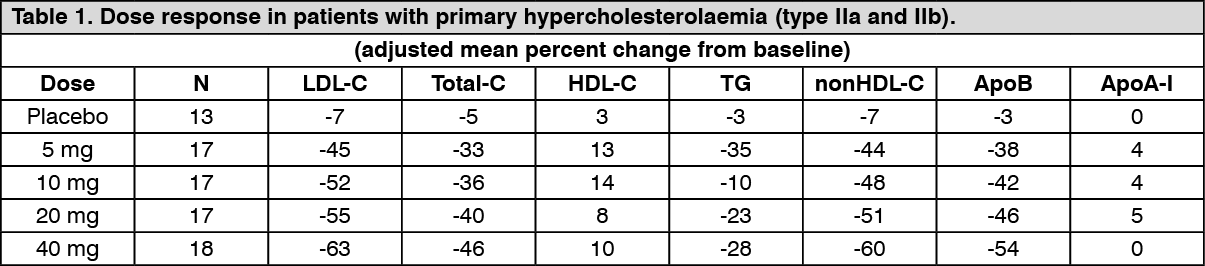
Pharmacodynamic effects: Rosuvastatin reduces elevated LDL-cholesterol, total cholesterol and triglycerides and increases HDL-cholesterol. It also lowers ApoB, nonHDL-C, VLDL-C, VLDL-TG and increases ApoA-I (see Table 1). Rosuvastatin also lowers the LDL-C/HDL-C, total C/HDL-C and nonHDL-C/HDL-C and the ApoB/ApoA-I ratios. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA therapeutic effect is obtained within 1 week following treatment initiation and 90% of maximum response is achieved in 2 weeks. The maximum response is usually achieved by 4 weeks and is maintained after that.
Ezetimibe: Ezetimibe is in a new class of lipid-lowering compounds that selectively inhibit the intestinal absorption of cholesterol and related plant sterols. Ezetimibe is orally active, and has a mechanism of action that differs from other classes of cholesterol-reducing compounds (e.g. statins, bile acid sequestrants [resins], fibric acid derivatives, and plant stanols). The molecular target of ezetimibe is the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1), which is responsible for the intestinal uptake of cholesterol and phytosterols.
Ezetimibe localises at the brush border of the small intestine and inhibits the absorption of cholesterol, leading to a decrease in the delivery of intestinal cholesterol to the liver; statins reduce cholesterol synthesis in the liver and together these distinct mechanisms provide complementary cholesterol reduction. In a 2-week clinical study in 18 hypercholesterolaemic patients, Ezetimibe inhibited intestinal cholesterol absorption by 54%, compared with placebo.
A series of preclinical studies was performed to determine the selectivity of ezetimibe for inhibiting cholesterol absorption. Ezetimibe inhibited the absorption of [14C]-cholesterol with no effect on the absorption of triglycerides, fatty acids, bile acids, progesterone, ethinyl estradiol, or fat soluble vitamins A and D.
Epidemiologic studies have established that cardiovascular morbidity and mortality vary directly with the level of total-C and LDL-C and inversely with the level of HDL-C. Administration of ezetimibe with a statin is effective in reducing the risk of cardiovascular events in patients with coronary heart disease and ACS event history.
Pharmacokinetics: Rosuvastatin and ezetimibe combination therapy: Concomitant use of 10 mg rosuvastatin and 10 mg ezetimibe resulted in a 1.2 fold increase in AUC of rosuvastatin in hypercholesterolaemic subjects. A pharmacodynamic interaction, in terms of adverse effects, between rosuvastatin and ezetimibe cannot be ruled out.
Rosuvastatin: Absorption: Maximum rosuvastatin plasma concentrations are achieved approximately 5 hours after oral administration. The absolute bioavailability is approximately 20%.
Distribution: Rosuvastatin is taken up extensively by the liver, which is the primary site of cholesterol synthesis and LDL-C clearance. The volume of distribution of rosuvastatin is approximately 134 L. Approximately 90% of rosuvastatin is bound to plasma proteins, mainly to albumin.
Biotransformation: Rosuvastatin undergoes limited metabolism (approximately 10%). In vitro metabolism studies using human hepatocytes indicate that rosuvastatin is a poor substrate for cytochrome P450-based metabolism. CYP2C9 was the principal isoenzyme involved, with 2C19, 3A4 and 2D6 involved to a lesser extent. The main metabolites identified are the N-desmethyl- and lactone metabolites. The N-desmethyl metabolite is approximately 50% less active than rosuvastatin whereas the lactone form is considered clinically inactive. Rosuvastatin accounts for greater than 90% of the circulating HMG-CoA reductase inhibitor activity.
Elimination: Approximately 90% of the rosuvastatin dose is excreted unchanged in the faeces (consisting of absorbed and non-absorbed active substance) and the remaining part is excreted in urine. Approximately 5% is excreted unchanged in urine. The plasma elimination half-life is approximately 19 hours. The elimination half-life does not increase at higher doses. The geometric mean plasma clearance is approximately 50 litres/hour (coefficient of variation 21.7%). As with other HMG-CoA reductase inhibitors, the hepatic uptake of rosuvastatin involves the membrane transporter OATP-C. This transporter is important in the hepatic elimination of rosuvastatin.
Linearity: Systemic exposure of rosuvastatin increases in proportion to dose. There are no changes in pharmacokinetic parameters following multiple daily doses.
Special populations: Age and sex: There was no clinically relevant effect of age or sex on the pharmacokinetics of rosuvastatin in adults. The exposure in children and adolescents with heterozygous familial hypercholesterolaemia appears to be similar to or lower than that in adult patients with dyslipidaemia (see "Paediatric population" as follows).
Race: Pharmacokinetic studies show an approximate 2-fold elevation in median AUC and Cmax in Asian subjects, including subjects of Japanese, Chinese, Malay and Indian ancestry, compared with Caucasians; Asian-Indians show an approximate 1.3-fold elevation in median AUC and Cmax.
A population pharmacokinetic analysis revealed no clinically relevant differences in pharmacokinetics between Caucasian and Black groups.
Renal insufficiency: In a study in subjects with varying degrees of renal impairment, mild to moderate renal disease had no influence on plasma concentration of rosuvastatin or the N-desmethyl metabolite. Subjects with severe impairment (CrCl <30 ml/min) had a 3-fold increase in plasma concentration and a 9-fold increase in the N-desmethyl metabolite concentration compared to healthy volunteers. Steady-state plasma concentrations of rosuvastatin in subjects undergoing haemodialysis were 50% greater compared to healthy volunteers.
Hepatic insufficiency: In a study with subjects with varying degrees of hepatic impairment there was no evidence of increased exposure to rosuvastatin in subjects with Child-Pugh scores of 7 or below. However, two subjects with Child-Pugh scores of 8 and 9 showed an increase in systemic exposure of at least 2-fold compared to subjects with lower Child-Pugh scores.
There is no experience in subjects with Child-Pugh scores above 9.
Genetic polymorphisms: Disposition of HMG-CoA reductase inhibitors, including rosuvastatin, involves OATP1B1 and BCRP transporter proteins. In patients with SLCO1B1 (OATP1B1) and/or ABCG2 (BCRP) genetic polymorphisms there is a risk of increased rosuvastatin exposure. Individual polymorphisms of SLCO1B1 c.521CC and ABCG2 c.421AA are associated with a higher rosuvastatin exposure (AUC) compared to the SLCO1B1 c.521TT or ABCG2 c.421CC genotypes. This specific genotyping is not established in clinical practice, but for patients who are known to have these types of polymorphisms, a lower daily dose of LYPSTAPLUS is recommended.
Paediatric population: Two pharmacokinetic studies with rosuvastatin (given as tablets) in paediatric patients with heterozygous familial hypercholesterolaemia 10-17 or 6-17 years of age (total of 214 patients) demonstrated that exposure in paediatric patients appears comparable to or lower than that in adult patients. Rosuvastatin exposure was predictable with respect to dose and time over a 2-year period.
Ezetimibe: Absorption: After oral administration, ezetimibe is rapidly absorbed and extensively conjugated to a pharmacologically-active phenolic glucuronide (ezetimibe-glucuronide). Mean maximum plasma concentrations (Cmax) occur within 1 to 2 hours for ezetimibe-glucuronide and 4 to 12 hours for ezetimibe. The absolute bioavailability of ezetimibe cannot be determined as the compound is virtually insoluble in aqueous media suitable for injection.
Concomitant food administration (high fat or non-fat meals) had no effect on the oral bioavailability of ezetimibe. Ezetimibe can be administered with or without food.
Distribution: Ezetimibe and ezetimibe-glucuronide are bound 99.7% and 88 to 92% to human plasma proteins, respectively.
Biotransformation: Ezetimibe is metabolised primarily in the small intestine and liver via glucuronide conjugation (a phase II reaction) with subsequent biliary excretion. Minimal oxidative metabolism (a phase I reaction) has been observed in all species evaluated. Ezetimibe and ezetimibe-glucuronide are the major drug-derived compounds detected in plasma, constituting approximately 10 to 20% and 80 to 90% of the total drug in plasma, respectively. Both ezetimibe and ezetimibe-glucuronide are slowly eliminated from plasma with evidence of significant enterohepatic recycling. The half-life for ezetimibe and ezetimibe-glucuronide is approximately 22 hours.
Elimination: Following oral administration of 14C-ezetimibe (20 mg) to human subjects, total ezetimibe accounted for approximately 93% of the total radioactivity in plasma. Approximately 78% and 11% of the administered radioactivity were recovered in the faeces and urine, respectively, over a 10-day collection period. After 48 hours, there were no detectable levels of radioactivity in the plasma.
Special populations: Age and sex: Plasma concentrations for total ezetimibe are about 2-fold higher in the elderly (≥65 years) than in the young (18 to 45 years). LDL-C reduction and safety profile are comparable between elderly and young subjects treated with ezetimibe. Therefore, no dosage adjustment is necessary in the elderly. Plasma concentrations for total ezetimibe are slightly higher (approximately 20%) in women than in men. LDL-C reduction and safety profile are comparable between men and women treated with ezetimibe. Therefore, no dosage adjustment is necessary on the basis of gender.
Renal insufficiency: After a single 10 mg dose of ezetimibe in patients with severe renal disease (n=8; mean CrCl ≤30 ml/min/1.73m2), the mean AUC for total ezetimibe was increased approximately 1.5-fold, compared to healthy subjects (n=9). This result is not considered clinically significant. No dosage adjustment is necessary for renally impaired patients.
An additional patient in this study (post-renal transplant and receiving multiple medications, including ciclosporin) had a 12-fold greater exposure to total ezetimibe.
Hepatic insufficiency: After a single 10 mg dose of ezetimibe, the mean AUC for total ezetimibe was increased approximately 1.7-fold in patients with mild hepatic insufficiency (Child Pugh score 5 or 6), compared to healthy subjects. In a 14-day, multiple-dose study (10 mg daily) in patients with moderate hepatic insufficiency (Child Pugh score 7 to 9), the mean AUC for total ezetimibe was increased approximately 4-fold on Day 1 and Day 14 compared to healthy subjects. No dosage adjustment is necessary for patients with mild hepatic insufficiency. Due to the unknown effects of the increased exposure to ezetimibe in patients with moderate or severe (Child Pugh score >9) hepatic insufficiency, LYPSTAPLUS is not recommended in these patients (see Precautions).
Paediatric population: The pharmacokinetics of ezetimibe are similar between children ≥6 years and adults. Pharmacokinetic data in the paediatric population <6 years of age are not available. Clinical experience in paediatric and adolescent patients includes patients with HoFH, HeFH, or sitosterolaemia.
Toxicology: Preclinical safety data: In co-administration studies with ezetimibe and statins the toxic effects observed were essentially those typically associated with statins. Some of the toxic effects were more pronounced than observed during treatment with statins alone. This is attributed to pharmacokinetic and pharmacodynamic interactions in co-administration therapy. No such interactions occurred in the clinical studies.
Myopathies occurred in rats only after exposure to doses that were several times higher than the human therapeutic dose (approximately 20 times the AUC level for statins and 500 to 2,000 times the AUC level for the active metabolites).
In a series of in vivo and in vitro assays ezetimibe, given alone or co-administered with statins, exhibited no genotoxic potential. Long-term carcinogenicity tests on ezetimibe were negative.
The co-administration of ezetimibe and statins was not teratogenic in rats. In pregnant rabbits a small number of skeletal deformities (fused thoracic and caudal vertebrae, reduced number of caudal vertebrae) were observed.
Rosuvastatin: Preclinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, genotoxicity and carcinogenicity potential. Specific tests for effects on hERG have not been evaluated. Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to clinical exposure levels were as follows: in repeated-dose toxicity studies histopathologic liver changes likely due to the pharmacologic action of rosuvastatin were observed in mouse, rat, and to a lesser extent with effects in the gall bladder in dogs, but not in monkeys. In addition, testicular toxicity was observed in monkeys and dogs at higher dosages. Reproductive toxicity was evident in rats, with reduced litter sizes, litter weight and pup survival observed at maternally toxic doses, where systemic exposures were several times above the therapeutic exposure level.
Ezetimibe: Animal studies on the chronic toxicity of ezetimibe identified no target organs for toxic effects. In dogs treated for four weeks with ezetimibe (≥0.03 mg/kg/day) the cholesterol concentration in the cystic bile was increased by a factor of 2.5 to 3.5. However, in a one-year study on dogs given doses of up to 300mg/kg/day no increased incidence of cholelithiasis or other hepatobiliary effects were observed. The significance of these data for humans is not known. A lithogenic risk associated with the therapeutic use of ezetimibe cannot be ruled out.
Ezetimibe had no effect on the fertility of male or female rats, nor was it found to be teratogenic in rats or rabbits, nor did it affect prenatal or postnatal development. Ezetimibe crossed the placental barrier in pregnant rats and rabbits given multiple doses of 1,000 mg/kg/day. The co-administration of ezetimibe with lovastatin resulted in embryolethal effects.