


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Olaparib is a potent inhibitor of human poly (ADP ribose) polymerase enzymes (PARP 1, PARP 2, and PARP 3), and has been shown to inhibit the growth of selected tumour cell lines in vitro and tumour growth in vivo either as a standalone treatment or in combination with established chemotherapies.
PARPs are required for the efficient repair of DNA single strand breaks and an important aspect of PARP-induced repair requires that after chromatin modification, PARP auto-modifies itself and dissociates from the DNA to facilitate access for base excision repair (BER) enzymes. When olaparib is bound to the active site of DNA-associated PARP it prevents the dissociation of PARP and traps it on the DNA, thus blocking repair. In replicating cells, this also leads to the formation of DNA double-strand breaks (DSBs) when replication forks meet the PARP DNA adducts. In normal cells, homologous recombination repair (HRR) pathway, which requires functional BRCA1 and 2 genes (Hard capsule), is effective at repairing these DNA DSBs. After a number of rounds of replication, genomic instability can reach insupportable levels and result in cancer cell death, as cancer cells already have a high DNA damage load relative to normal cells.
In BRCA-deficient in vivo models, olaparib given after platinum treatment resulted in a delay in tumour progression and an increase in overall survival compared to platinum treatment alone that correlated with the period of olaparib maintenance treatment.
Hard capsule: In the absence of functional BRCA1 or 2, DNA DSBs cannot be repaired via HRR. Instead, alternative and error-prone pathways are activated, such as the non-homologous end joining (NHEJ) pathway, leading to increased genomic instability.
FC tablet: In HR-deficient cancer cells, the repair of these DNA DSBs is impaired. Cancer cells can become HR deficient due to inactivation of genes with a direct or indirect role in HRR, such as BRCA1/2, ATM, CDK12 and others. Instead, alternative and error-prone pathways are activated, such as the classical non-homologous end joining (NHEJ) pathway, leading to increased genomic instability. In the absence of deleterious mutations in key HRR genes, HRR pathway may be compromised by other mechanisms, although the causative aberrancy and penetrance are not fully elucidated. Absence of fully functional HRR pathway is one of the key determinants of platinum sensitivity in ovarian and other cancers.
Clinical data: Hard capsule: Platinum Sensitive Relapsed (PSR) Ovarian Cancer: The safety and efficacy of Lynparza as a maintenance therapy in the treatment of PSR ovarian, fallopian tube or primary peritoneal cancer patients, following treatment with two or more platinum containing regimens, were studied in a Phase II randomised, double blind placebo controlled trial (Study 19). The study compared the efficacy of olaparib maintenance treatment taken to progression with no maintenance treatment in 265 (136 olaparib and 129 placebo) PSR patients who were in response (CR [complete response] or PR [partial response]) following completion of platinum containing chemotherapy. The primary endpoint was PFS based on investigator assessment using RECIST 1.0. Secondary efficacy endpoints included OS (overall survival), DCR (disease control rate) defined as confirmed CR/PR + SD (stable disease), HRQoL (health related quality of life), and disease related symptoms. Exploratory analyses of time to first subsequent therapy or death (TFST) and time to second subsequent therapy or death (TSST) were also performed.
Only PSR patients who were in response following completion of platinum based chemotherapy and whose disease had recurred >6 months after completion of prior penultimate platinum based chemotherapy were enrolled. Patients could not have received prior olaparib or other PARP inhibitor treatment. Patients could have received prior bevacizumab, except in the regimen immediately prior to randomisation.
The study met its primary objective of demonstrating a statistically significant and clinically relevant improvement in PFS for olaparib maintenance monotherapy compared with placebo in the overall population (hazard ratio [HR] 0.35; 95% CI 0.25-0.49; p<0.00001); median 8.4 months olaparib vs 4.8 months placebo). At the final analysis (data cut off [DCO] 9 May 2016) for OS at 79% maturity, the HR comparing olaparib with placebo was 0.73 (95% CI 0.55-0.95; p=0.02138 [did not meet pre-specified significance level of <0.0095]; median 29.8 months olaparib versus 27.8 months placebo).
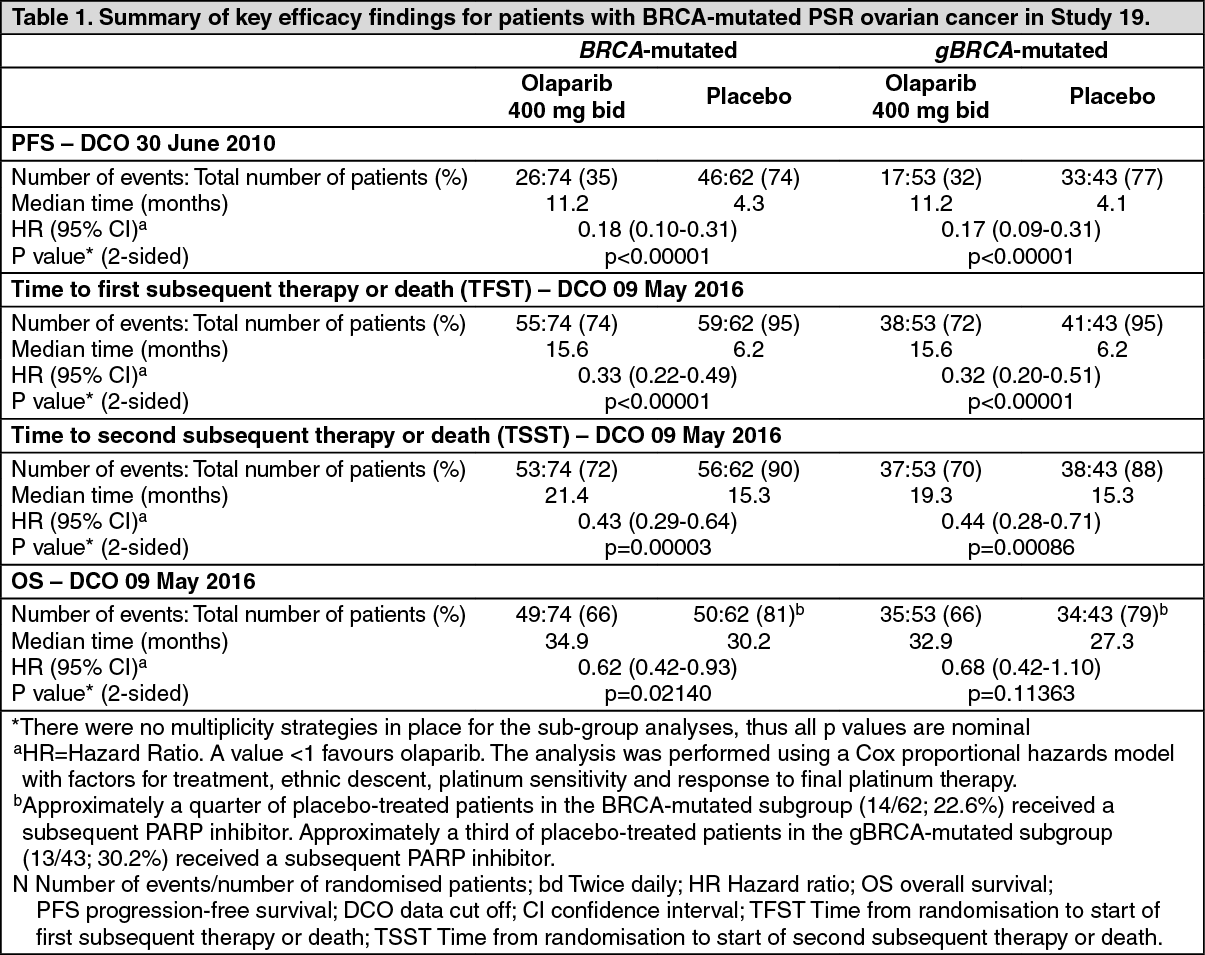
Preplanned subgroup analysis identified patients with BRCA-mutated ovarian cancer (n=136, 51.3%) as the subgroup that derived the greatest clinical benefit from olaparib maintenance monotherapy. There were no multiplicity strategies in place for the sub-group analyses.
In BRCA-mutated patients (n=136) there was a statistically significant improvement in PFS, TFST, and TSST. The median PFS improvement was 6.9 months over placebo (HR 0.18; 95% CI 0.10-0.31; p<0.00001; median 11.2 months vs 4.3 months). At the final analysis (data cut off [DCO] 9 May 2016) the time from randomisation to start of first subsequent therapy or death (TFST) was 9.4 months longer for olaparib treated patients (HR 0.33; 95% CI 0.22-0.49; p<0.00001; median 15.6 months versus 6.2 months). The time from randomisation to start of second subsequent therapy or death (TSST) was 6.1 months longer for olaparib treated patients (HR 0.43; 95% CI 0.29-0.64; p=0.00003; median 21.4 months versus 15.3 months. For the secondary endpoint of OS, the HR for olaparib vs. placebo was 0.62 (95% CI 0.42-0.93; p=0.02140; median 34.9 months versus 30.2 months). In the olaparib-treated group, 28.4% of patients remained on treatment for ≥2 years and 14.9% for ≥5 years. In the placebo-treated group, 8.1% of patients remained on treatment for ≥2 years and 1.6% for ≥5 years.
A summary of key efficacy findings for patients with BRCA-mutated PSR ovarian cancer in Study 19 is presented in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWithin the BRCA-mutated population the disease control rate at 24 weeks was 57% and 24% for patients in the olaparib and placebo groups, respectively.
No statistically significant differences were observed between treatment groups in patient reported symptoms or HRQoL.
Effect on the QT Interval: There is no clinically relevant effect of olaparib on cardiac repolarisation (as evaluated by an effect on the QT interval) following 300 mg twice daily multiple dosing of the Lynparza tablet formulation.
FC tablet: Maintenance treatment of newly diagnosed advanced ovarian cancer: SOLO1 Study in newly diagnosed advanced patients with a BRCA mutation: SOLO1 was a Phase III randomised, double-blind, placebo-controlled, multicentre trial that compared the efficacy of Lynparza maintenance treatment (300 mg [2 x 150 mg tablets] twice daily) with placebo in advanced (FIGO Stage III-IV) high-grade serous or endometrioid BRCA-mutated (BRCAm) ovarian cancer. The study randomised 391 patients (2:1 randomisation: 260 olaparib and 131 placebo) who were in response (CR [complete response] or PR [partial response]) following completion of first-line platinum-containing chemotherapy. Patients were stratified by response to first-line platinum chemotherapy (CR or PR). Treatment was continued for 2 years or until progression of the underlying disease. For patients who remained in complete clinical response (i.e. no radiological evidence of disease), the maximum duration of treatment was 2 years; however, patients who had evidence of disease that remained stable (i.e. no evidence of disease progression) could continue to receive Lynparza beyond 2 years.
Patients with BRCA mutations were identified either from germline testing in blood via a local test or central test (i.e. Myriad Integrated BRACAnalysis test, Myriad BRACAnalysis CDx, China BGI test) or from testing a tumour sample using a local test. The BRCAm status of all patients was confirmed where possible using the Myriad Integrated BRACAnalysis test, the Myriad BRACAnalysis CDx or the Foundation Medicine FoundationOne CDx Clinical Trial Assay.
There were 389 patients who were germline BRCAm and 2 who were somatic BRCAm in SOLO1.
Demographic and baseline characteristics were generally well balanced between the olaparib and placebo treatment arms. Median age was 53 years in both arms. Ovarian cancer was the primary tumour in 85% of the patients. The most common histological type was serous (96%), endometrioid histology was reported in 2% of the patients. Most patients were ECOG performance status 0 (78%). All patients had received first-line platinum-based therapy; response to prior platinum chemotherapy was complete in 82% and partial in 18% of the patients. Ninety three percent (93%) of patients were randomised within 8 weeks of their last dose of platinum-based chemotherapy.
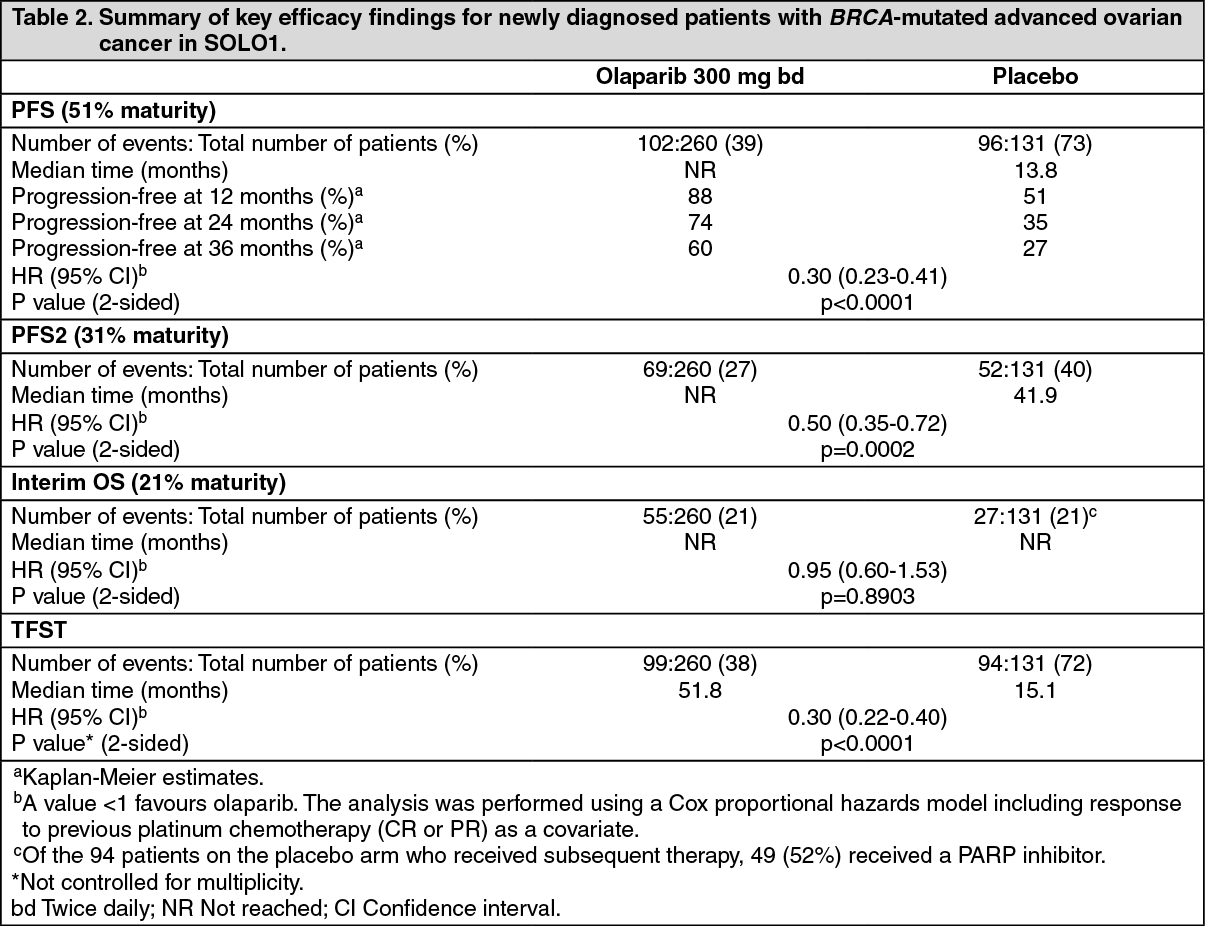
The primary endpoint was progression-free survival (PFS), defined as time from randomisation to progression determined by investigator assessment using modified Response Evaluation Criteria in Solid Tumors (RECIST) 1.1, or death. Secondary efficacy endpoints included time from randomisation to second progression or death (PFS2), overall survival (OS), time from randomisation to first subsequent anti-cancer therapy or death (TFST) and health related quality of life (HRQoL). Patients had tumour assessments at baseline and every 12 weeks for 3 years, and then every 24 weeks relative to the date of randomisation, until objective radiological disease progression.
The study demonstrated a clinically relevant and statistically significant improvement in investigator assessed PFS for olaparib compared to placebo, with a hazard ratio (HR) of 0.30 (95% CI 0.23-0.41; p< 0.0001; the median was not reached for olaparib versus 13.8 months for placebo). Based on Kaplan-Meier estimates, the proportion of patients that were progression free at 12, 24 and 36 months were 88%, 74%, and 60% for olaparib versus 51%, 35% and 27% for placebo; the median follow-up time was 41 months for both the olaparib and placebo treatment arms. The investigator assessment of PFS was supported with a blinded independent central radiological (BICR) review of PFS (HR 0.28; 95% CI 0.20-0.39; p< 0.0001; median not reached for olaparib vs. 14.1 months for placebo). A clinically meaningful and statistically significant improvement in PFS2 was also observed with a HR of 0.50 (95% CI 0.35-0.72; p=0.0002; median not reached for olaparib vs. 41.9 months for placebo) indicating that the benefit observed with olaparib continued to be evident even with the use of subsequent therapies (see Table 2).
At the time of PFS analysis, interim OS data were immature with events in 82/391 (21%) patients (HR 0.95; 95% CI 0.60-1.53; p=0.8903; medians not reached). A clinically meaningful and statistically significant improvement in TFST was observed for olaparib-treated patients (Table 2). (See Table 2 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image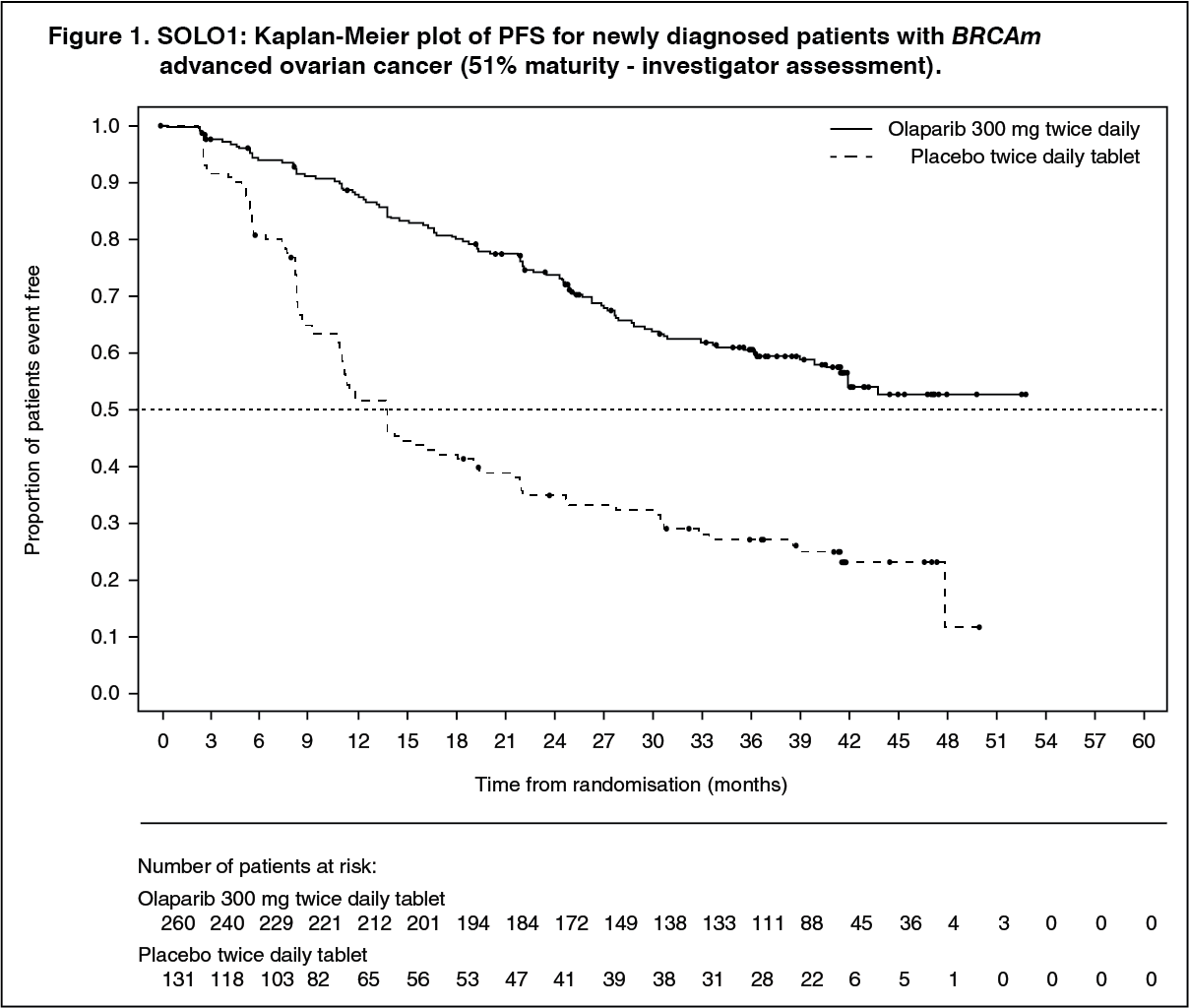 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThere was no decrease in HRQoL from baseline for olaparib-treated patients over the 24-month treatment period and no clinically relevant differences in HRQoL compared with placebo-treated patients as assessed by the change from baseline in the Trial Outcome Index (TOI) of the Functional Assessment of Cancer Therapy - Ovarian (FACT-O).
Platinum-sensitive relapsed (PSR) ovarian cancer: The efficacy of Lynparza in the maintenance treatment setting in platinum-sensitive relapsed (PSR) ovarian, fallopian tube or primary peritoneal cancer is supported by 2 randomised, double-blind, placebo-controlled trials in patients with PSR and BRCA-mutated disease (SOLO2) and in patients with PSR disease (Study 19). In both studies, PSR patients who were in response following completion of platinum-based chemotherapy and whose disease had recurred >6 months after completion of penultimate platinum-based chemotherapy were enrolled. Patients could not have received prior olaparib or other PARP inhibitor treatment. Patients could have received prior bevacizumab, except in the regimen immediately prior to randomisation. Patients with BRCA mutations were identified either from germline testing in blood via a local test or the Myriad CLIA Integrated BRACAnalysis test or from testing a tumour sample using a local test or a test performed by Foundation Medicine.
In addition, the efficacy of Lynparza in the maintenance treatment setting in non-gBRCAm PSR ovarian, fallopian tube or primary peritoneal cancer was also assessed in a single-arm, multicentre study (OPINION).
SOLO2 Study in PSR patients with a BRCA mutation: The study compared the efficacy of Lynparza maintenance treatment (300 mg [2 x 150 mg tablets] twice daily) taken to progression with placebo treatment in 295 patients with high-grade serous or endometrioid PSR ovarian cancer (2:1 randomisation: 196 olaparib and 99 placebo) who were in response (CR or PR) following completion of platinum-containing chemotherapy. All patients had evidence of germline BRCA mutation (gBRCAm) at baseline.
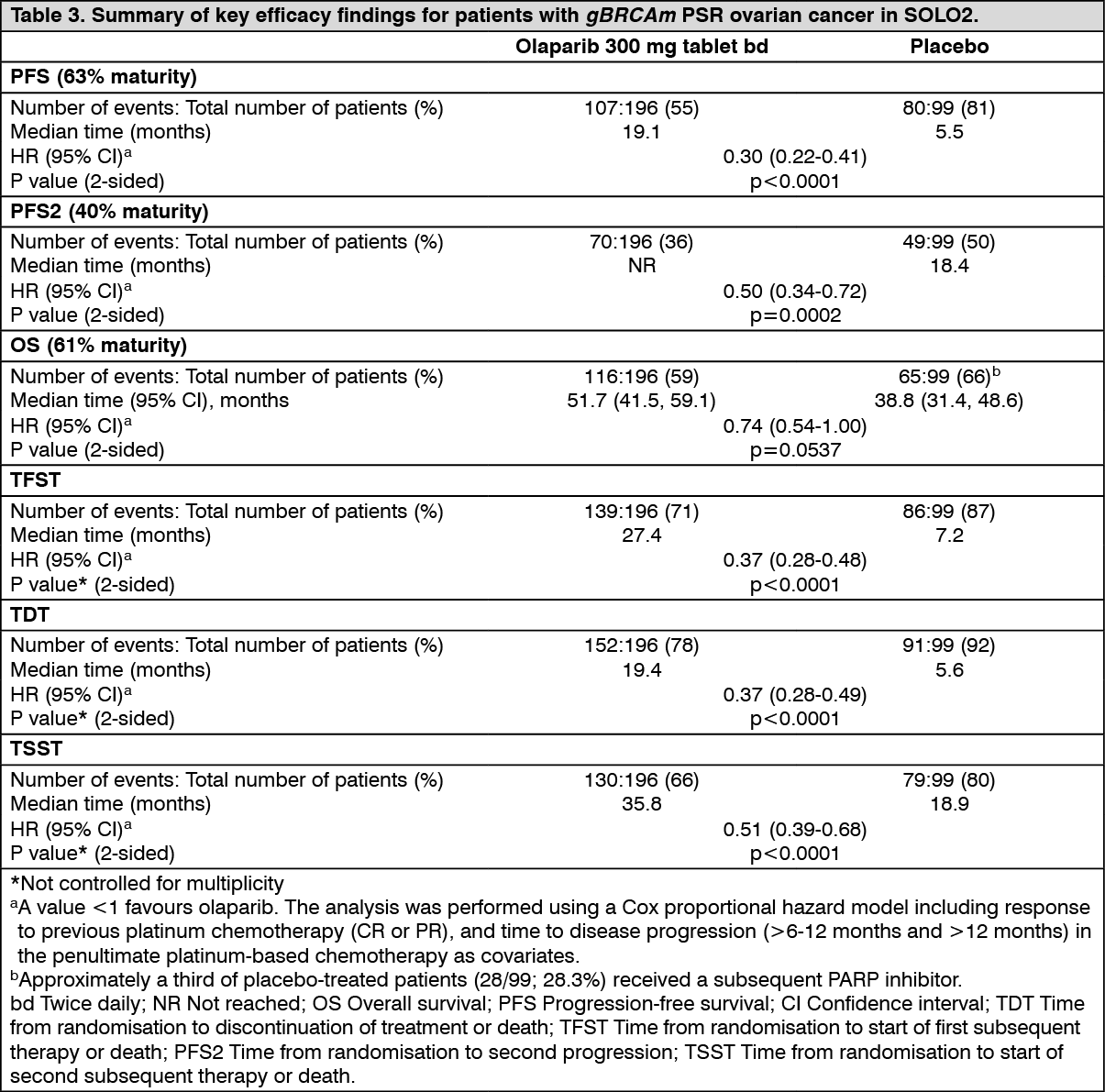
The primary endpoint was PFS determined by investigator assessment using RECIST 1.1. Secondary efficacy endpoints included PFS2; OS, TDT, TFST, TSST; and HRQoL.
The study met its primary objective demonstrating a clinically meaningful and statistically significant improvement in investigator assessed PFS for olaparib compared with placebo with a HR of 0.30 (95% CI 0.22-0.41; p<0.0001; median 19.1 months for olaparib vs. 5.5 months for placebo). The investigator assessment of PFS was supported with a blinded independent central radiological review of PFS (HR 0.25; 95% CI 0.18-0.35; p<0.0001; median 30.2 months for olaparib vs. 5.5 months for placebo). At 2 years, 43% olaparib-treated patients remained progression-free compared with only 15% placebo-treated patients. A clinically meaningful and statistically significant improvement in PFS2 was also observed with a HR of 0.50 (95% CI 0.34-0.72; p=0.0002; median not reached for olaparib vs. 18.4 months for placebo) indicating that the benefit observed with olaparib continued to be evident even with the use of subsequent therapies. At the final analysis of OS (61% maturity) the HR was 0.74 (95% CI 0.54-1.00; p=0.0537; median 51.7 months for olaparib vs 38.8 months for placebo) which did not reach statistical significance.
Clinically meaningful and statistically significant improvements in TDT, TFST and TSST were also observed for olaparib-treated patients (Table 3).
A summary of key efficacy findings for patients with gBRCAm PSR ovarian cancer in SOLO2 is presented in Table 3. (See Table 3, Figures 2 and 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image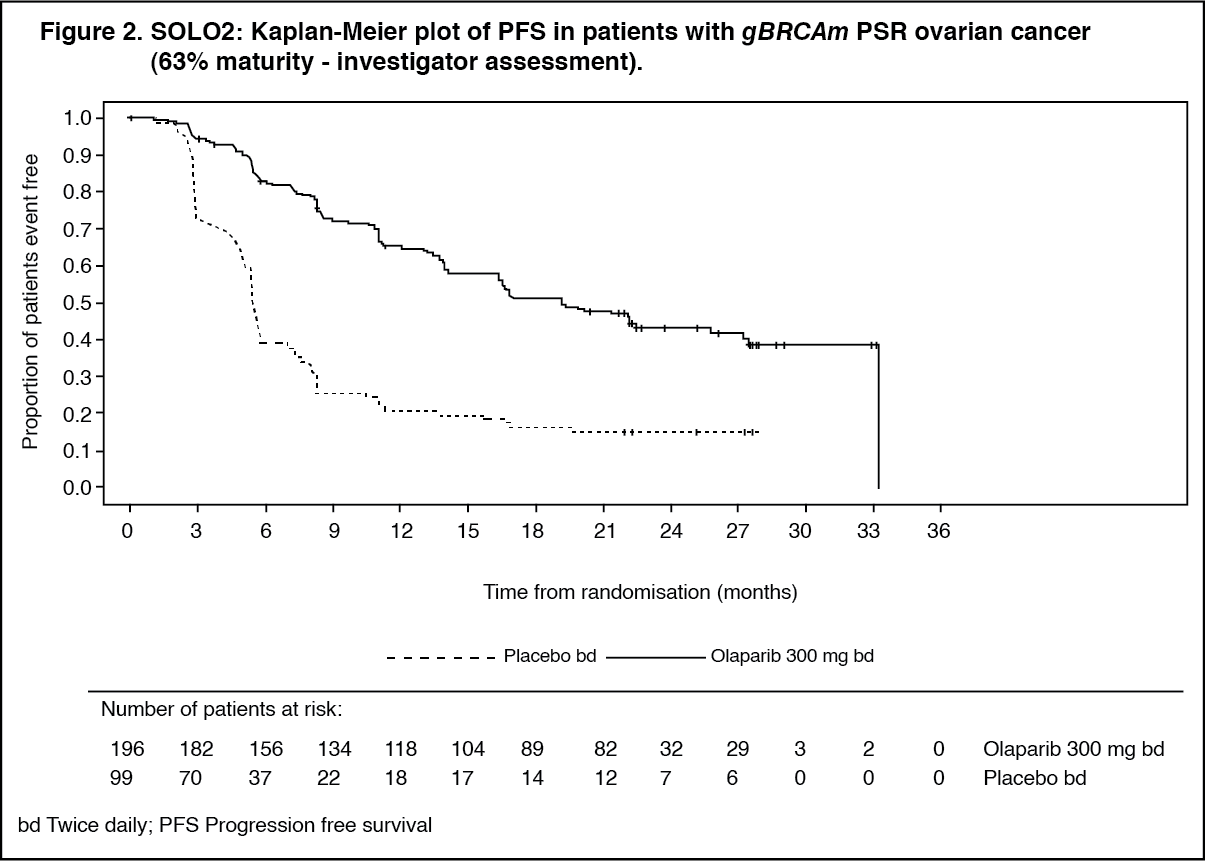 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image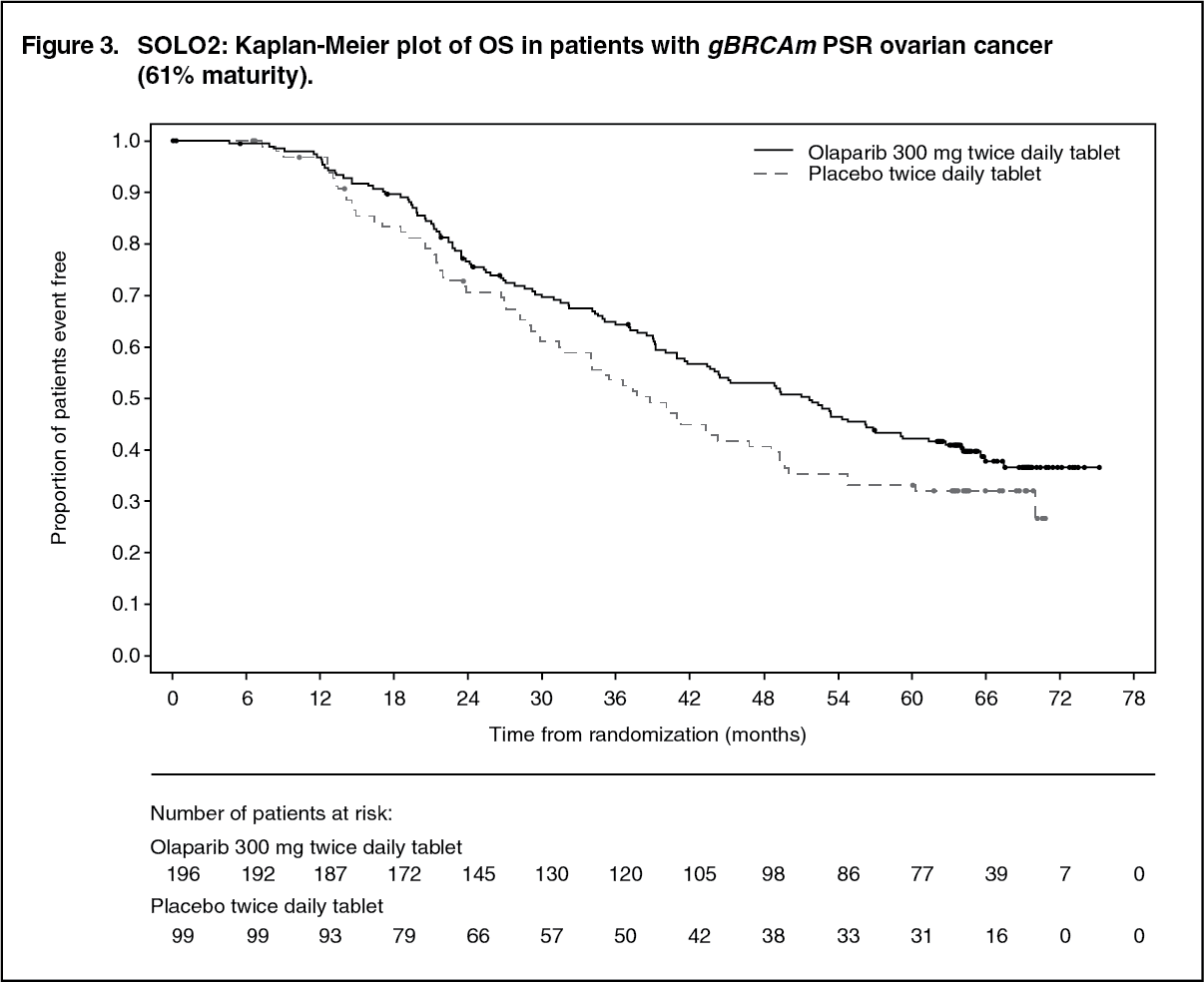 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThere was no difference between olaparib and placebo treatment groups in HRQoL as assessed by the change from baseline in the Trial Outcome Index (TOI) of the Functional Assessment of Cancer Therapy - Ovarian (FACT-O) over 12 months (estimated difference - 0.03; 95% CI: -2.191, 2.2126; p=0.9765).
Study 19 in PSR patients: The study compared the efficacy of Lynparza capsule maintenance treatment (400 mg [8 x 50 mg capsules] twice daily) taken to progression with placebo treatment in 265 (136 olaparib and 129 placebo) PSR patients who were in response (CR or PR) following completion of platinum-containing chemotherapy. The primary endpoint was PFS based on investigator assessment using RECIST 1.0. Secondary efficacy endpoints included OS, disease control rate (DCR), HRQoL, and disease related symptoms. Exploratory analyses TFST and TSST were also performed.
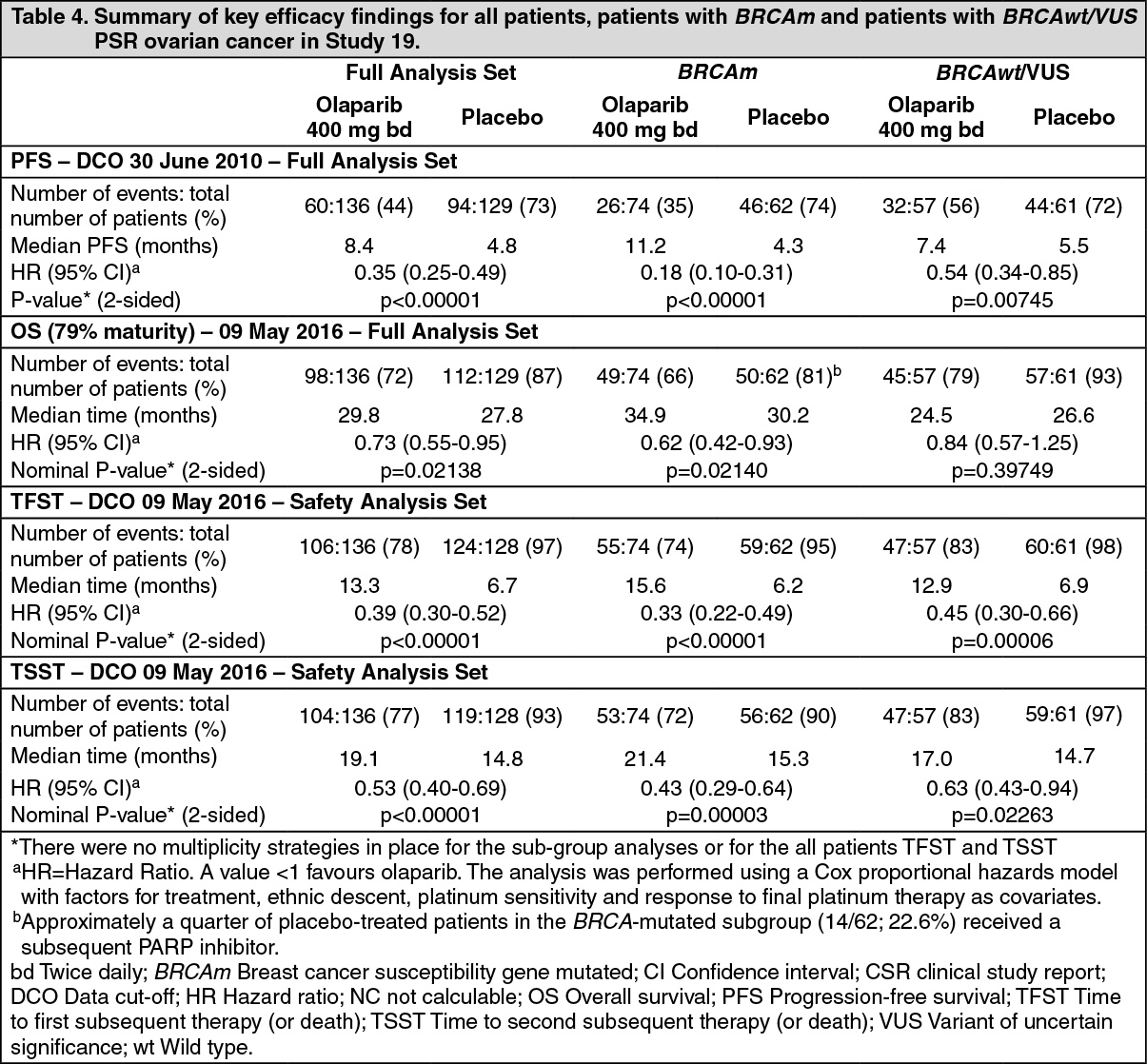
The study met its primary objective demonstrating a statistically significant and clinically relevant improvement in PFS for olaparib compared with placebo with a HR of 0.35 (95% CI 0.25-0.49; p<0.00001; median 8.4 months for olaparib vs. 4.8 months for placebo). At the final analysis (data cut off [DCO] 9 May 2016) for OS at 79% maturity, the HR comparing olaparib with placebo was 0.73 (95% CI 0.55-0.95; p=0.02138 [did not meet pre-specified significance level of <0.0095]; median 29.8 months for olaparib vs. 27.8 months for placebo). TFST and TSST were also longer for olaparib-treated patients (Table 4).
Preplanned subgroup analysis identified patients with BRCAm ovarian cancer (n=136, 51.3%) as the subgroup that derived the greatest clinical benefit from olaparib maintenance monotherapy. There were no multiplicity strategies in place for the sub-group analyses. In BRCAm patients the HR for PFS improvement was 0.18 (95% CI 0.10-0.31; p<0.00001; median 11.2 months for olaparib vs. 4.3 months for placebo). For the secondary endpoint of OS, the HR for olaparib vs. placebo was 0.62 (95% CI 0.42-0.93; p=0.02140; median 34.9 months for olaparib vs. 30.2 months for placebo). In the olaparib-treated group, 28.4% of patients remained on treatment for ≥2 years and 14.9% for ≥5 years. In the placebo-treated group, 8.1% of patients remained on treatment for ≥2 years and 1.6% for ≥5 years. TFST and TSST were also longer for olaparib-treated patients (Table 4).
A summary of key efficacy findings for all patients, patients with BRCAm and patients with BRCAwt/VUS PSR ovarian cancer in Study 19 is presented in Table 4. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWithin the overall population, the DCR at 24 weeks was 53% and 25% for patients in the olaparib and placebo groups, respectively and in the BRCAm population was 57% and 24% for patients in the olaparib and placebo groups, respectively.
No statistically significant differences were observed between treatment groups in patient reported symptoms or HRQoL.
OPINION study in non-gBRCAm PSR ovarian cancer patients: OPINION was a single arm, multicentre study that investigated olaparib (300 mg [2 x 150 mg tablets] twice daily) as a maintenance treatment in patients with PSR high grade serous or endometrioid ovarian cancer following 2 or more lines of platinum-based chemotherapy and who did not have a known deleterious or suspected deleterious gBRCA mutation. Patients whose disease was in response (CR or PR) following completion of platinum-based chemotherapy were enrolled. A total of 279 patients were enrolled and received olaparib treatment until disease progression or unacceptable toxicity.
Based on central retrospective germline blood testing, 90.7% of patients were confirmed with a non-gBRCAm status, 2.2% with gBRCAm status and 7.2% of patients had no confirmatory gBRCA result. Based on retrospective central blood and tumour testing, 9.7% of patients were identified as sBRCAm and 13.3% of patients had a HRD/BRCA status of unknown.
The primary endpoint was investigator-assessed PFS according to modified RECIST v1.1. Secondary endpoints included OS.
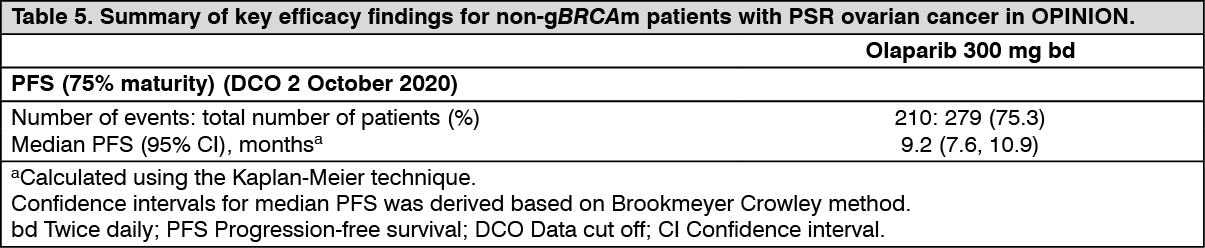
Olaparib when used as maintenance therapy, demonstrated clinical activity in patients with non-gBRCAm PSR ovarian cancer. At the time of primary PFS analysis, the OS data were 30% mature.
A summary of the key efficacy findings in patients with non-gBRCAm PSR ovarian cancer in OPINION is presented in Table 5. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFirst-line maintenance treatment of HRD positive advanced ovarian cancer: PAOLA-1 study in newly-diagnosed advanced ovarian cancer patients: PAOLA-1 was a Phase III randomised, double-blind, placebo-controlled, multicentre trial that compared the efficacy of Lynparza (300 mg [2 x 150 mg tablets] twice daily) in combination with bevacizumab (15 mg/kg of body weight given once every 3 weeks as an intravenous infusion) compared with placebo plus bevacizumab for the maintenance treatment of newly-diagnosed advanced (FIGO Stage III-IV) high-grade epithelial ovarian, fallopian tube or primary peritoneal cancer.
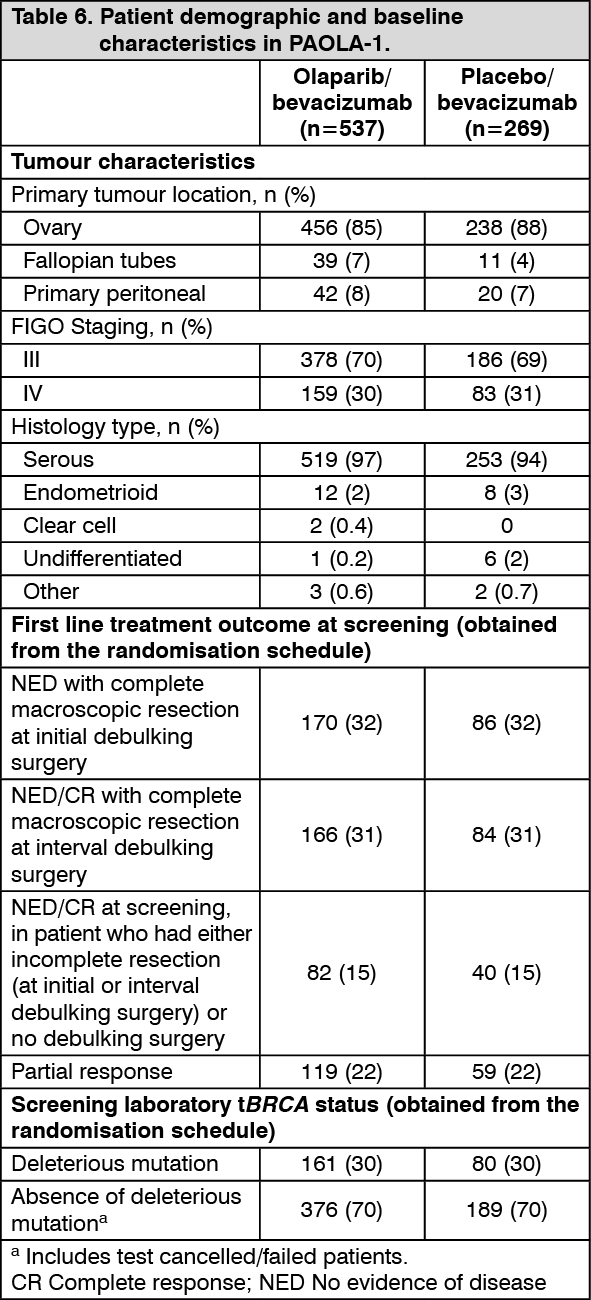
The study randomised 806 patients (2:1 randomisation: 537 olaparib/bevacizumab: 269 placebo/bevacizumab) who had no evidence of disease (NED) due to complete surgical resection, or who were in complete response (CR), or partial response (PR) following completion of first-line platinum-containing chemotherapy and bevacizumab. Patients were stratified by first-line treatment outcome (timing and outcome of cytoreductive surgery and response to platinum-based chemotherapy) and tBRCAm status, determined by prospective local testing. Patients continued bevacizumab in the maintenance setting and started treatment with Lynparza after a minimum of 3 weeks and up to a maximum of 9 weeks following completion of their last dose of chemotherapy. Treatment with Lynparza was continued for up to 2 years or until progression of the underlying disease. Patients who in the opinion of the treating physician could derive further benefit from continuous treatment could be treated beyond 2 years. Treatment with bevacizumab was for a total of up to 15 months, including the period given with chemotherapy and given as maintenance.
Demographic and baseline characteristics were well balanced between both the study and placebo arms in the Intention to Treat (ITT) population and also in the HRD-positive subgroup. The median age of patients was 61 years overall. Most patients in both arms were ECOG performance status 0 (70%). Ovarian cancer was the primary tumour in 86% of the patients. The most common histological type was serous (96%) and endometrioid histology was reported in 2% of the patients. Most patients were diagnosed in FIGO stage IIIC (63%). All patients had received first-line platinum-based therapy and bevacizumab. Patients were not restricted by the surgical outcome with 65% having complete cytoreduction at initial or interval debulking surgery and 35% having residual macroscopic disease. In the HRD-positive subgroup, 65% of patients had complete cytoreduction and 35% of patients had residual macroscopic disease.
The median duration of treatment with Lynparza was 17.3 months and 15.6 months for placebo. The median duration of bevacizumab post-randomisation was 11.0 months on the Lynparza arm and 10.4 months on the placebo arm. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe primary endpoint was progression-free survival (PFS), defined as time from randomisation to progression determined by investigator assessment using modified Response Evaluation Criteria in Solid Tumors (RECIST) 1.1, or death. Secondary efficacy endpoints included time from randomisation to second progression or death (PFS2), overall survival (OS), time from randomisation to first subsequent anti-cancer therapy or death (TFST) and health related quality of life (HRQoL). Patients had RECIST 1.1 tumour assessments at baseline and every 24 weeks (CT/MRI at 12 weeks if clinical or CA 125 progression) for up to 42 months or until objective radiological disease progression.
The study met its primary end-point in the ITT population demonstrating a statistically significant improvement in investigator assessed PFS for olaparib/bevacizumab compared to placebo/bevacizumab (HR 0.59, 95% CI 0.49-0.72, p<0.0001 with a median of 22.1 months for olaparib/bevacizumab vs 16.6 months for placebo/bevacizumab). Final analysis of PFS2 (DCO 22 March 2020, 53% maturity) in the overall population was statistically significant (HR 0.78, 95% CI 0.64-0.95, p=0.0125 with a median of 36.5 months for olaparib/bevacizumab vs 32.6 months for placebo/bevacizumab).
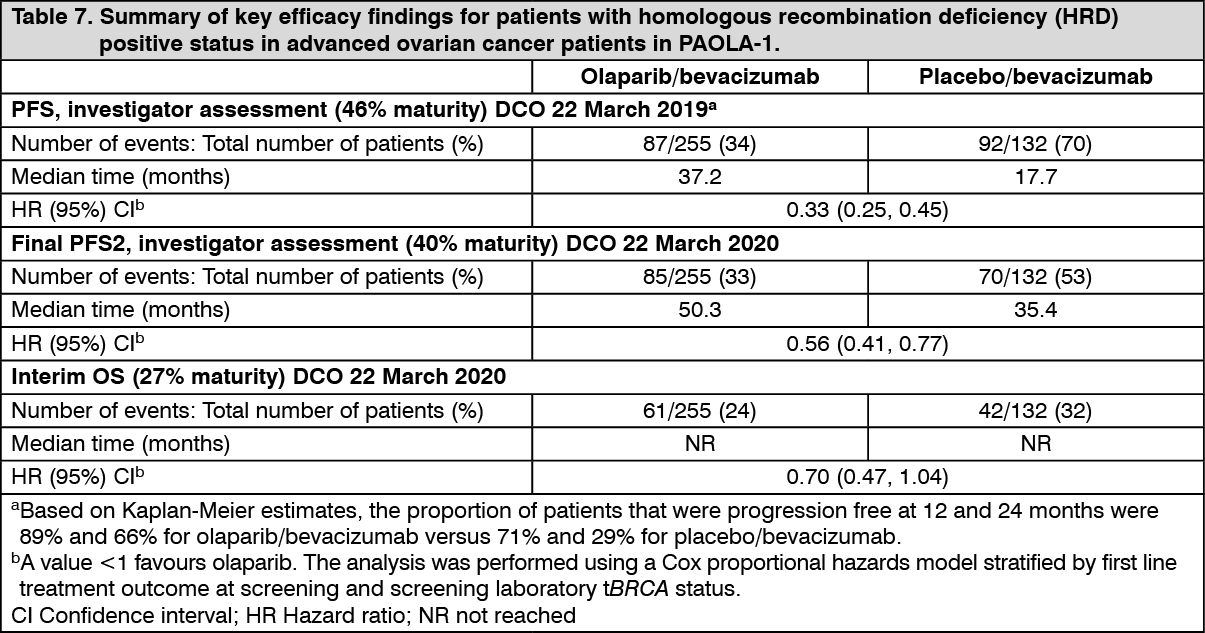
Efficacy results from a biomarker subgroup analysis of 387 patients with HRD-positive tumours, identified post-randomization, who received olaparib/bevacizumab (n=255) or placebo/bevacizumab (n=132), are summarized in Table 7 and Figure 4. Results from a blinded independent review of PFS were consistent. Overall survival data in this subpopulation were immature with 27% deaths. (See Table 7 and Figure 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image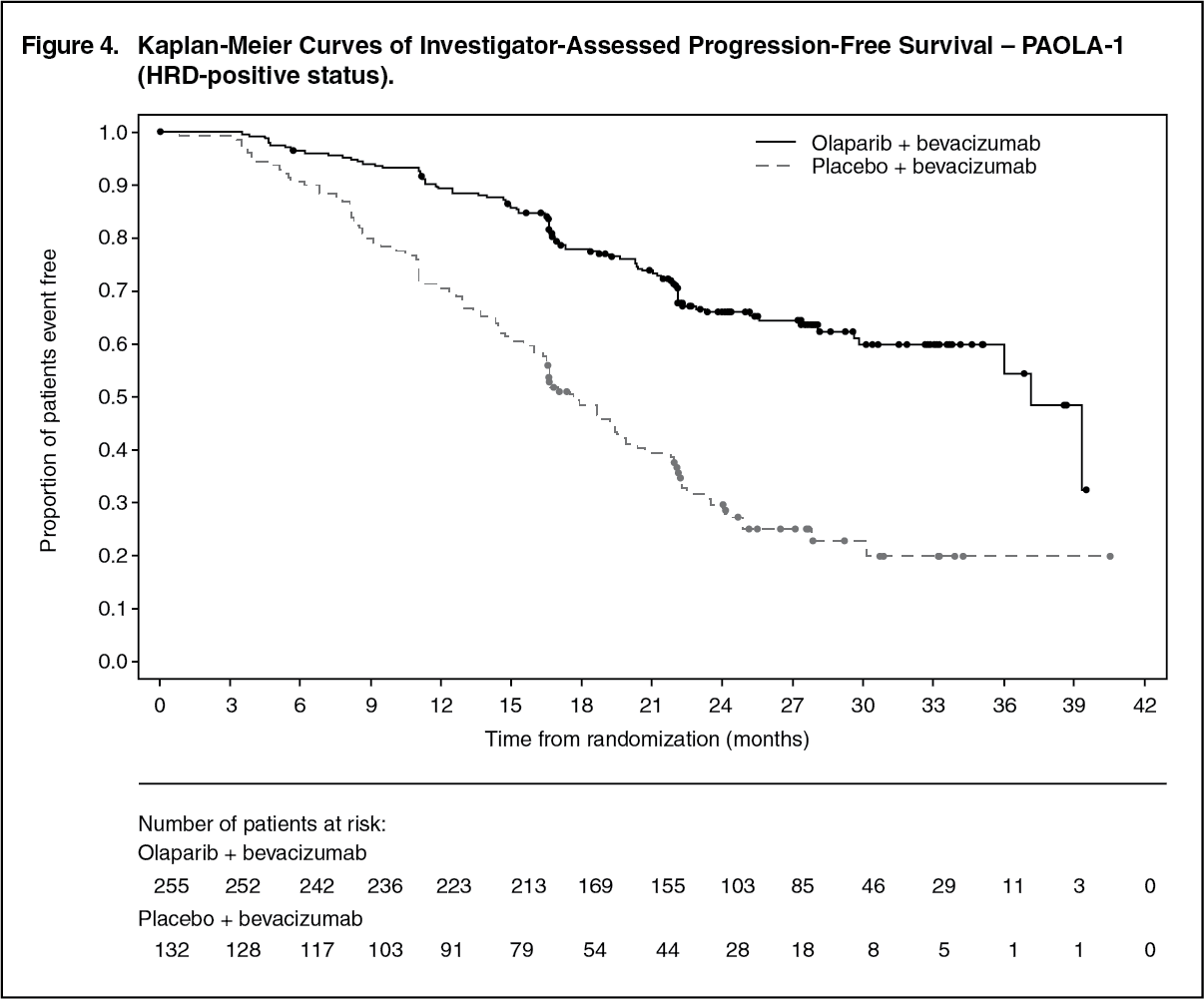 Click on icon to see table/diagram/image
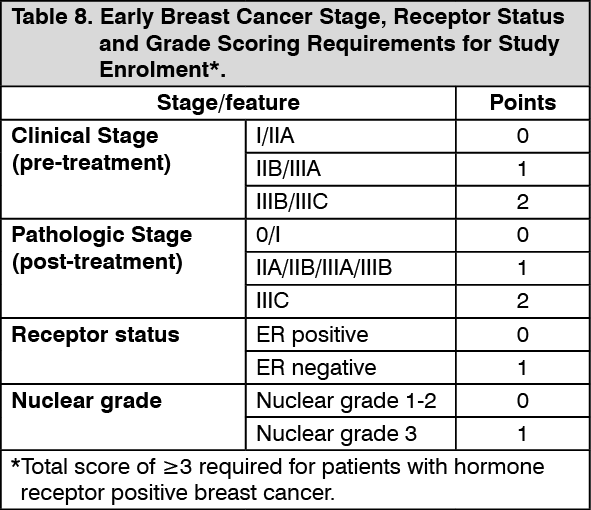
Click on icon to see table/diagram/imageAdjuvant treatment of germline BRCA-mutated HER2-negative high risk early breast cancer: OlympiA study in HER2-negative high risk early breast cancer patients with a germline BRCA mutation: OlympiA was a Phase III randomised, double-blind, parallel group, placebo-controlled, multicentre study to assess the efficacy and safety of olaparib (300 mg [2 x 150 mg tablets] twice daily) vs placebo as adjuvant treatment in patients with germline BRCA1/2 mutations and HER2-negative high risk early breast cancer who had completed definitive local treatment and neoadjuvant or adjuvant chemotherapy. Patients were required to have completed at least 6 cycles of neoadjuvant or adjuvant chemotherapy containing anthracyclines, taxanes or both. Prior platinum for previous cancer (e.g. ovarian) or as adjuvant or neoadjuvant treatment for breast cancer was allowed. High risk early breast cancer patients were defined as follows: patients who received prior neoadjuvant chemotherapy: patients with either triple negative breast cancer (TNBC) or hormone receptor positive breast cancer must have had residual invasive cancer in the breast and/or the resected lymph nodes (non-pathologic complete response) at the time of surgery. Additionally, patients with hormone receptor positive breast cancer must have had a score of ≥3 based on pre-treatment clinical and post-treatment pathologic stage (CPS), estrogen receptor (ER) status and histologic grade as shown in Table 8. (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients who have received prior adjuvant chemotherapy: triple negative breast cancer (TNBC) patients must have had node positive disease or node negative disease with a ≥2cm primary tumor; ER and/or PgR positive, HER2-negative patients must have had ≥4 pathologically confirmed positive lymph nodes.
Patients were randomised in a 1:1 ratio to either olaparib (n=921) or placebo (n=915). Randomisation was stratified by hormone receptor status (ER and/or PgR positive/ HER2 negative versus TNBC), by prior neoadjuvant versus adjuvant chemotherapy, and by prior platinum use for breast cancer (yes versus no). Treatment was continued for 1 year, or until disease recurrence, or unacceptable toxicity.
The primary endpoint was invasive disease free survival (IDFS), defined as the time from randomisation to date of first recurrence, where recurrence is defined as loco-regional, distant recurrence, contralateral invasive breast cancer, new cancer or death from any cause. Secondary objectives included OS, distant disease free survival [(DDFS, defined as the time from randomisation until evidence of first distant recurrence of breast cancer or death from any cause), the incidence of new primary contralateral breast cancers (invasive and non-invasive), new primary ovarian cancer, new primary fallopian tube cancer and new primary peritoneal cancer], and patient reported outcomes using the FACIT-Fatigue and EORTC QLQ-C30 questionnaires.
Central testing with Myriad BRACAnalysis or local gBRCA testing, if available, was used to establish study eligibility. Patients enrolled based on local gBRCA test results provided a sample for retrospective confirmatory testing with BRACAnalysis (excluding patients enrolled in China). Out of 1836 patients enrolled into OlympiA, 1539 were confirmed as gBRCAm by Myriad BRACAnalysis, either prospectively or retrospectively.
Demographic and baseline characteristics were well balanced between the arms. The median age was 42 years. Sixty-seven percent (67%) of patients were White, 29% Asian and 2.6% Black. Two patients (0.2%) in the olaparib arm and four patients (0.4%) in the placebo arm were male. Sixty-one percent (61%) of patients were pre-menopausal. Eighty-nine percent (89%) of patients were ECOG performance status 0 and 11% ECOG PS 1. Eighty-two percent (82%) of patients had TNBC and 18% had hormone receptor-positive disease (defined as ER positive and/or PgR positive). Fifty percent (50%) of patients had received prior neoadjuvant and 50% received prior adjuvant chemotherapy. Ninety-four percent (94%) of patients received anthracycline and taxane. Twenty-six (26%) of patients overall had received prior platinum for breast cancer. In the olaparib and placebo arms, 87% and 92% of patients with HR positive disease were receiving concomitant endocrine therapy, respectively.
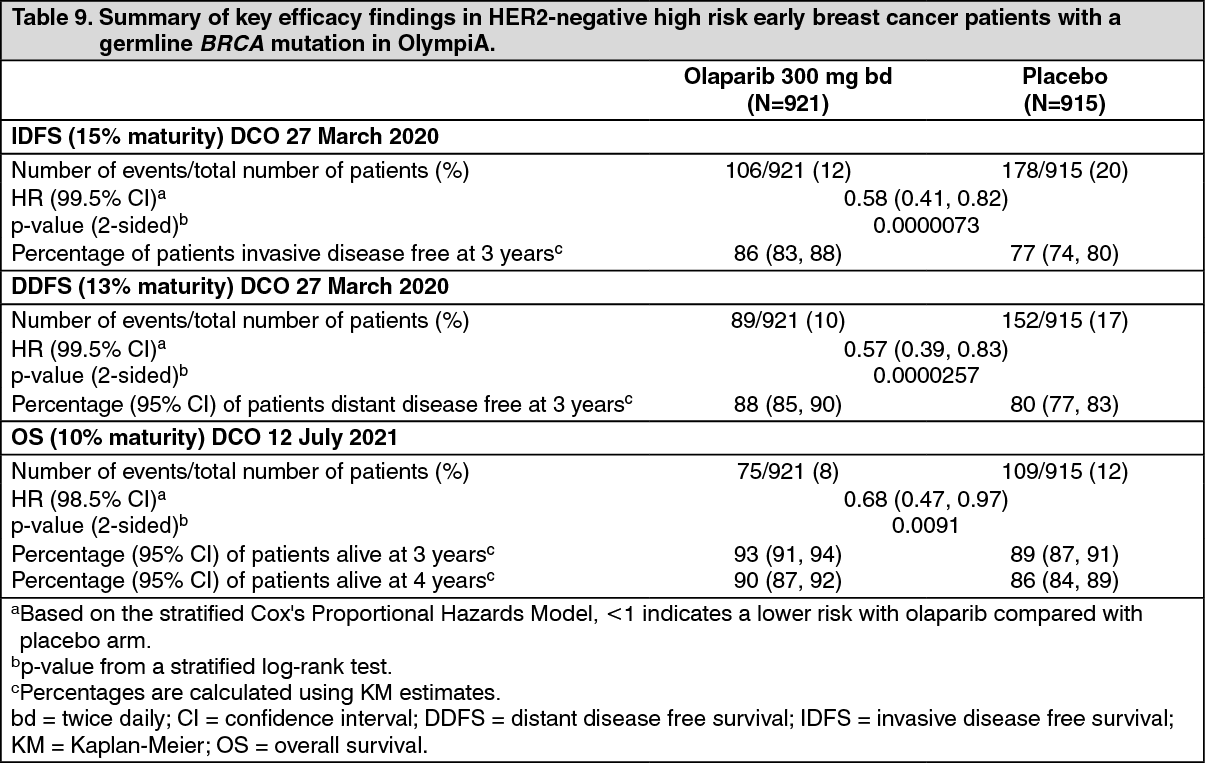
The study demonstrated a statistically significant improvement in IDFS in the olaparib arm compared with the placebo arm. Two hundred and eighty-four (284) patients had IDFS events, this represented 12% of patients in the olaparib arm (distant 8%, local/regional 1.4%, contralateral invasive breast cancer 0.9%, non-breast second primary malignancies 1.2%, death 0.2%) and 20% of patients in the placebo arm (distant 13%, local/regional 2.7%, contralateral invasive breast cancer 1.3%, non-breast second primary malignancies 2.3%, death 0%). A statistically significant improvement in DDFS in the olaparib arm compared with the placebo arm was also observed. At the next planned OS analysis, a statistically significant improvement in OS was observed in the olaparib arm compared with the placebo arm.
Efficacy results from the FAS are presented in Table 9 and for the subgroups by stratification factors in Table 10. (See Tables 9 and 10, Figures 5, 6 and 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image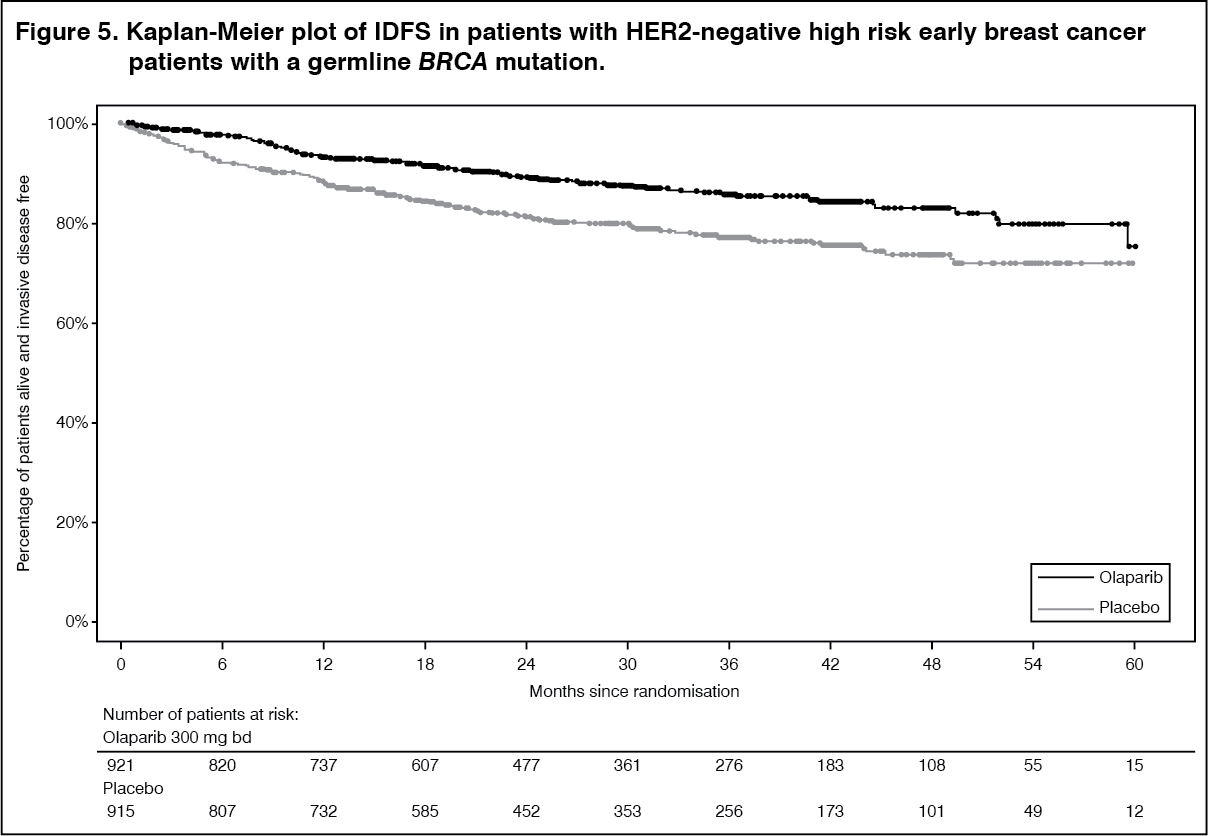 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image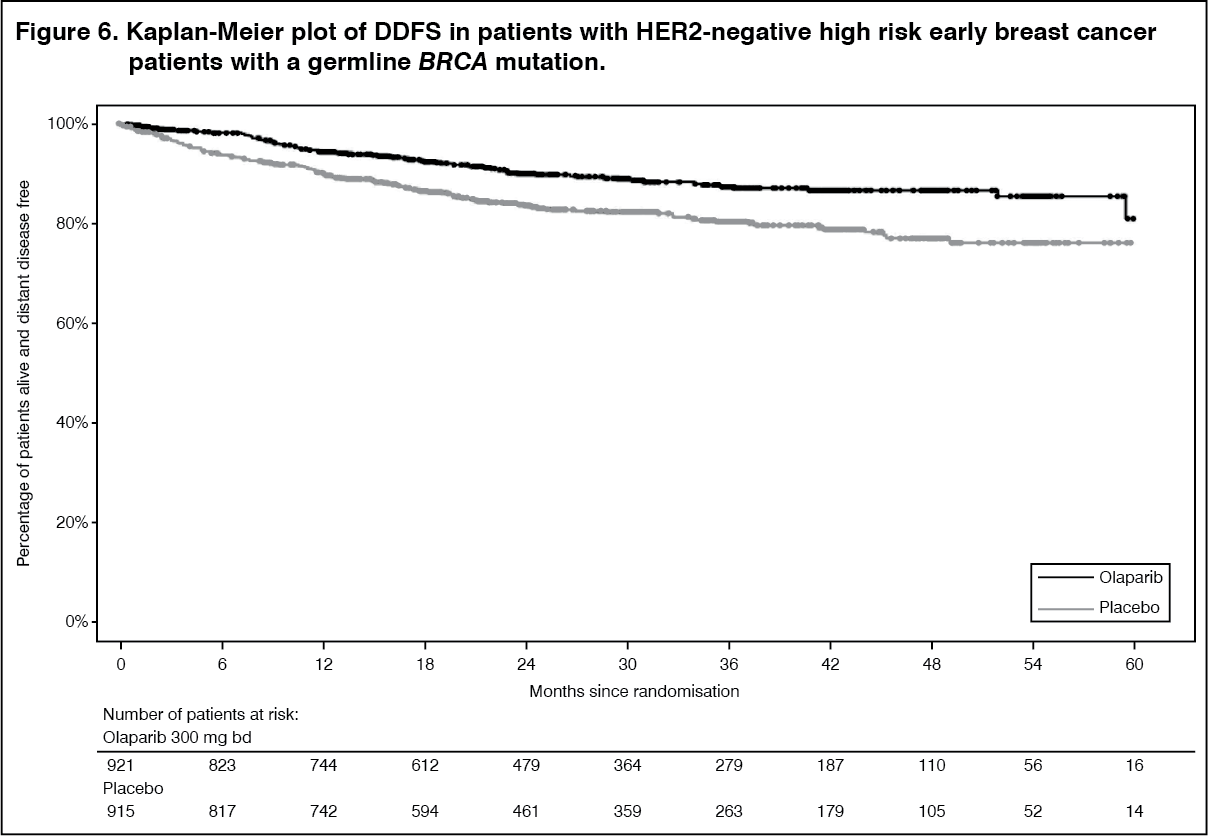 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image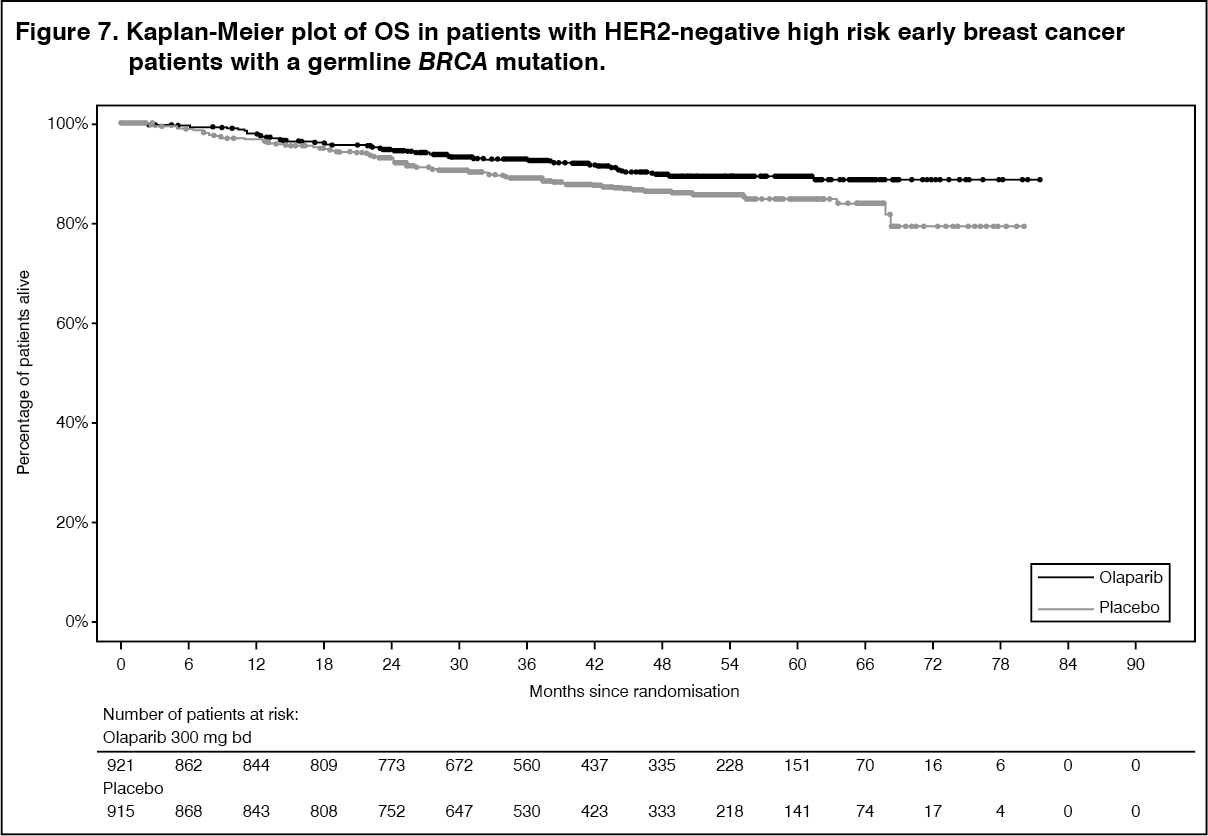 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image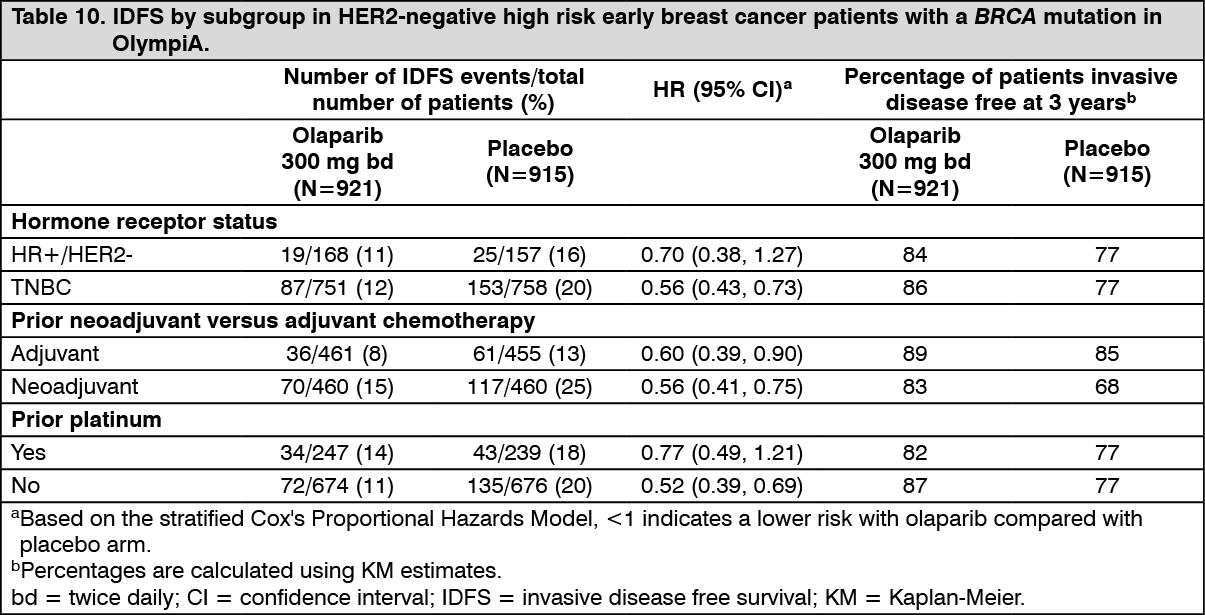 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe patient reported outcome assessments included the FACIT-Fatigue (to assess fatigue and its impact upon daily activities and function) which were completed at baseline prior to randomisation and at 6 months and 12 months after randomisation. A 3-point difference in FACIT-Fatigue score was predefined as clinically meaningful. Patient reported outcome data indicate no clinically meaningful differences among olaparib-treated patients as compared to placebo when measured using FACIT-Fatigue scale. These results should be interpreted cautiously as this secondary endpoint was not adjusted for multiplicity testing in the statistical analysis.
Germline BRCA-mutated HER2-negative metastatic breast cancer: OlympiAD in HER2-negative metastatic breast cancer patients with a gBRCA mutation: The study was a Phase III randomised, open-label, controlled trial that compared the efficacy of olaparib (300 mg [2 x 150 mg tablets] twice daily) taken to progression with a comparator arm of physician's choice of chemotherapy (capecitabine, eribulin or vinorelbine). In the study 302 patients with gBRCAm HER2-negative metastatic breast cancer who had previously received up to two lines of chemotherapy for the treatment of metastatic disease were randomised (2:1 randomisation: 205 olaparib and 97 comparator). Patients were stratified based on: receipt of prior chemotherapy regimens for metastatic breast cancer, oestrogen receptor (ER) and/or progesterone receptor (PgR) positive vs ER and PgR negative, prior platinum for breast cancer. The primary endpoint was PFS assessed by BICR using RECIST 1.1. Secondary endpoints included PFS2, OS, objective response rate (ORR) and HRQoL.
All patients had received prior treatment with anthracycline (unless contraindicated) and a taxane in either the neoadjuvant, adjuvant or metastatic setting. Prior therapy with platinum for metastatic breast cancer was allowed provided there had been no evidence of disease progression during platinum treatment. Prior therapy with platinum in the (neo)adjuvant setting was allowed provided the last dose was received at least 12 months prior to randomisation. Patients could not have received prior olaparib or other PARP inhibitor treatment. Patients with ER and/or PgR positive disease must have received and progressed on at least one endocrine therapy (adjuvant or metastatic) or had disease that the treating physician believed to be inappropriate for endocrine therapy. Patients had tumour assessments at baseline and every 6 weeks for the first 24 weeks, and then every 12 weeks relative to date of randomisation, until objective radiological disease progression.
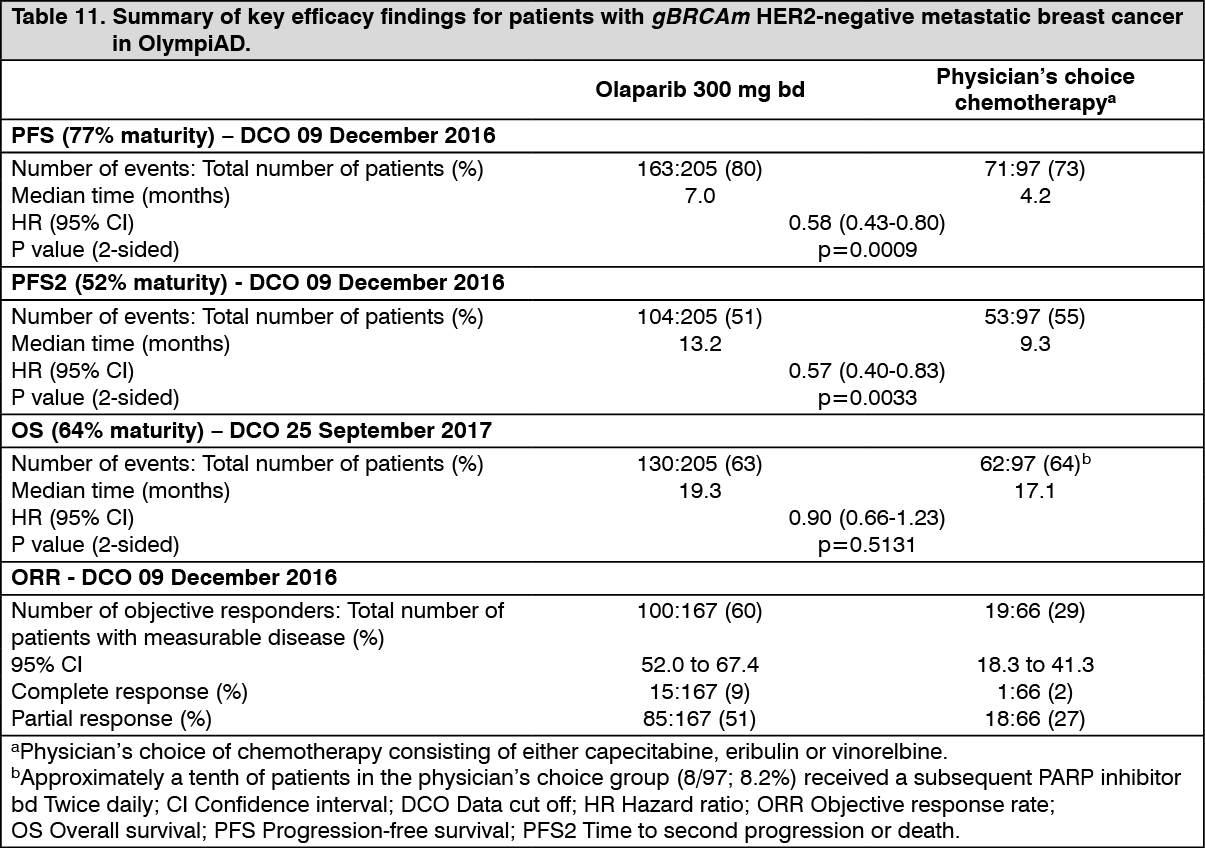
The study met its primary objective demonstrating a statistically significant and clinically meaningful improvement in PFS for olaparib-treated patients compared with those in the comparator arm with a HR of 0.58 (95% CI 0.43-0.80; p=0.0009; median 7.0 months for olaparib vs. 4.2 months for comparator) (Table 11).
A clinically meaningful and statistically significant improvement in PFS2 was also observed with a HR of 0.57 (95% CI 0.40-0.83; p=0.0033; median 13.2 months for olaparib vs 9.3 months for comparator) indicating that the benefit observed with olaparib continued to be evident even with the use of subsequent therapies. In the measurable disease patient population (77%), ORR in olaparib-treated patients was 60% (95% CI 52.0-67.4) and in patients who received comparator was 29% (95% CI 18.3-41.3). The median time to onset of response was 47 days for olaparib vs 45 days for comparator. The median duration of response was 6.4 months for olaparib vs 7.1 months for comparator. Overall survival was 64% mature at the time of the final OS analysis (DCO 25 September 2017). The OS HR comparing olaparib with comparator was 0.90 (95% CI 0.66-1.23; p=0.5131; median 19.3 months for olaparib vs. 17.1 months for comparator). The median follow-up time in censored patients was 25.3 months for olaparib vs 26.3 months for comparator.
Consistent results were observed across patient subgroups. (See Table 11 and Figure 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image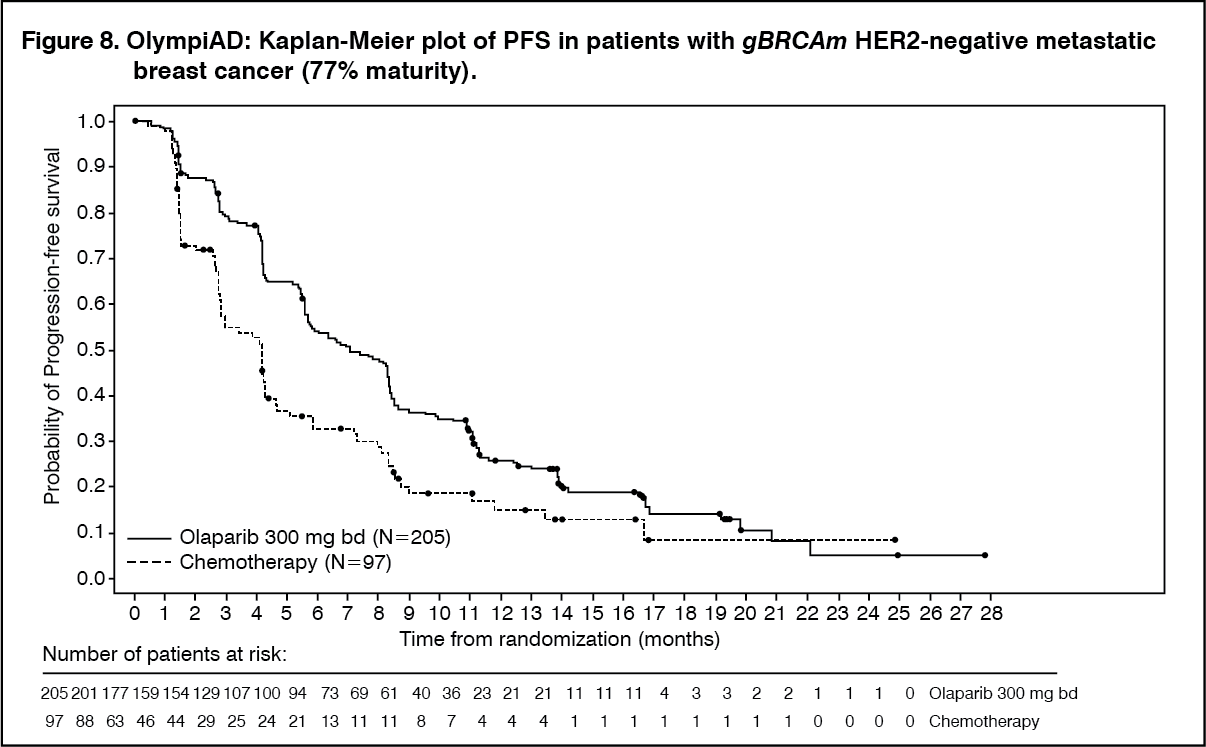 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA significant difference in global health status/QoL (assessed using the EORTC QLQ-C30 questionnaire which uses a 0-100 point scale) in favour of olaparib was observed (adjusted mean difference in change from baseline score was 7.5 points [95% CI: 2.48-12.44; p=0.0035]). Time to deterioration (≥10 points decrease from baseline) in global health status/QoL score was statistically significantly longer on the olaparib arm (HR 0.44; 95% CI: 0.25-0.77; p=0.0043; median not reached for olaparib vs. 15.3 months for comparator arm). Over the treatment period, the proportion of patients with clinically significant improvement (≥10 points increase from baseline) in global health status/QoL score was 33.7% (n=69) in the olaparib arm and 13.4% (n=13) in the comparator arm.
Maintenance following first-line treatment of germline BRCA-mutated metastatic adenocarcinoma of the pancreas: POLO was a Phase III, randomised, double-blind, placebo-controlled, multicentre trial that compared the efficacy of Lynparza maintenance treatment (300 mg [2 x 150 mg tablets] twice daily) with placebo in gBRCA-mutated metastatic adenocarcinoma of the pancreas. The study randomised 154 patients (3:2 randomisation: 92 olaparib and 62 placebo) whose disease had not progressed following at least 16 weeks of first-line platinum-based chemotherapy. There was no upper limit to the duration of chemotherapy received. After 16 weeks of continuous platinum-based chemotherapy, the platinum could be discontinued at any time for toxicity and the other agents continued; the patients were eligible for randomisation as long as there was no evidence of progression at any time during chemotherapy treatment. All toxicities from previous anti-cancer therapy must have been resolved to CTCAE grade 1, except for alopecia, grade 3 peripheral neuropathy and Hgb ≥ 9 g/dL. Lynparza treatment was continued until progression of the underlying disease.
Patients with germline BRCA mutations were identified from prior local testing results or by central testing using the Myriad BRACAnalysis or Myriad BRACAnalysis CDx test. The BRCAm status of all patients identified using prior local testing results was confirmed, where sent, using the Myriad BRACAnalysis or Myriad BRACAnalysis CDx test.
Demographic and baseline characteristics were generally well balanced between the olaparib and placebo arms. Median age was 57 years in both arms; 30% of patients in the olaparib arm were ≥ 65 years compared to 21% in the placebo arm. Fifty-eight per-cent (58%) of patients were male. Most patients were ECOG performance status 0 (67%). Ninety-six per-cent (96%) of patients were randomised within 8 weeks of their last dose of platinum-based chemotherapy. The median time from initiation of first-line platinum-based chemotherapy to randomisation was 5.8 months (range 3.4 to 33.4 months) and 49% of patients were in complete or partial response to their most recent platinum-based regimen.
The primary endpoint was progression-free survival (PFS), defined as time from randomisation to progression determined by BICR using modified Response Evaluation Criteria in Solid Tumors (RECIST) 1.1, or death. Secondary efficacy endpoints included overall survival (OS), time from randomisation to second progression or death (PFS2), time from randomisation to first subsequent anti-cancer therapy or death (TFST), time from randomisation to discontinuation of treatment or death (TDT), objective response rate (ORR), duration of response (DoR), response rate, time to response and health related quality of life (HRQoL). Patients had tumour assessments at baseline and every 8 weeks for 40 weeks, and then every 12 weeks relative to the date of randomisation, until objective radiological disease progression. For PFS, the median follow-up time for censored patients was 9.1 months in the olaparib arm and 3.8 months in the placebo arm. For OS, the median follow-up time for censored patients was 31.3 months in the olaparib arm and 23.9 months in the placebo arm.
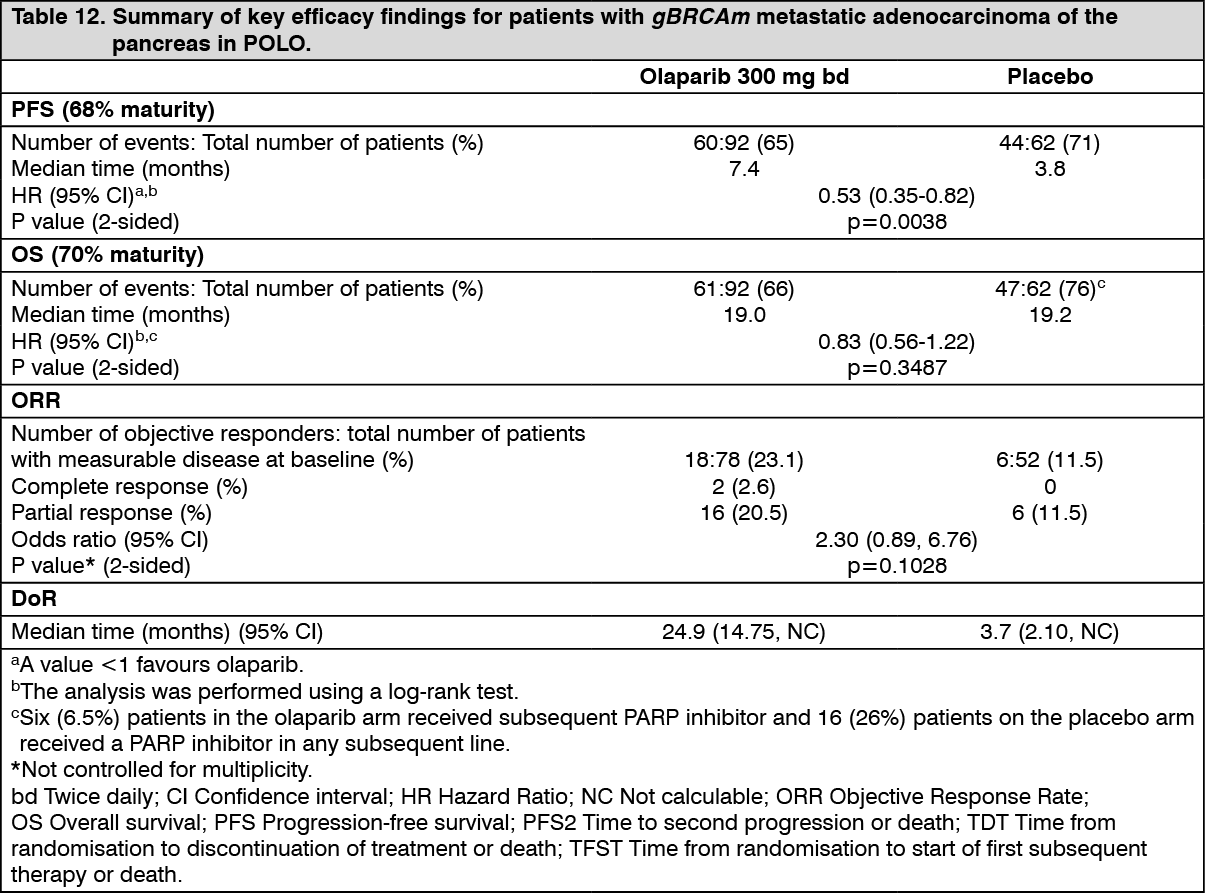
The study demonstrated a clinically meaningful and statistically significant improvement in PFS for olaparib compared to placebo, with a HR of 0.53 (95% CI 0.35 - 0.82; p=0.0038; the median was 7.4 months for olaparib vs 3.8 months for placebo). The sensitivity analysis of PFS by investigator assessment (HR 0.51; 95% CI 0.34 to 0.78; p=0.0017; median 6.3 months vs 3.7 months for olaparib vs placebo, respectively) was consistent with the PFS analysis by BICR. Based on Kaplan-Meier estimates, the proportion of patients that were alive and progression-free at 12, 24 and 36 months were 34%, 28% and 22% for olaparib vs 15%, 10% and 10% for placebo.
At the time of PFS analysis, the median DoR was longer in the olaparib arm (24.9 months) compared to the placebo arm (3.7 months), with a longer median time to onset of response (5.4 months for olaparib vs 3.6 months for placebo).
At the final analysis of OS (70% maturity) the HR for OS was 0.83 (95% CI 0.56 to 1.22; p=0.3487; median 19.0 months for olaparib vs 19.2 months for placebo) which did not reach statistical significance. The percentage of patients that were alive and in follow-up were 28% in the olaparib arm and 18% in the placebo arm.
At the time of final OS analysis, the HR for PFS2 (60% maturity, not controlled for multiplicity) was 0.66 (95% CI 0.42 - 1.02; p=0.0613) with a difference in median of 7.6 months in favour of olaparib (median 16.9 months for olaparib vs 9.3 months for placebo). The median TFST was 9.0 months in the olaparib arm and 5.4 months in the placebo arm; HR 0.44; 95% CI 0.30, 0.69; and p<0.0001 [nominal]. The median TDT was 7.5 months in the olaparib arm and 3.8 months in the placebo arm; HR 0.43; 95% CI 0.29, 0.63; and p<0.0001 [nominal]. (See Table 12, Figures 9 and 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image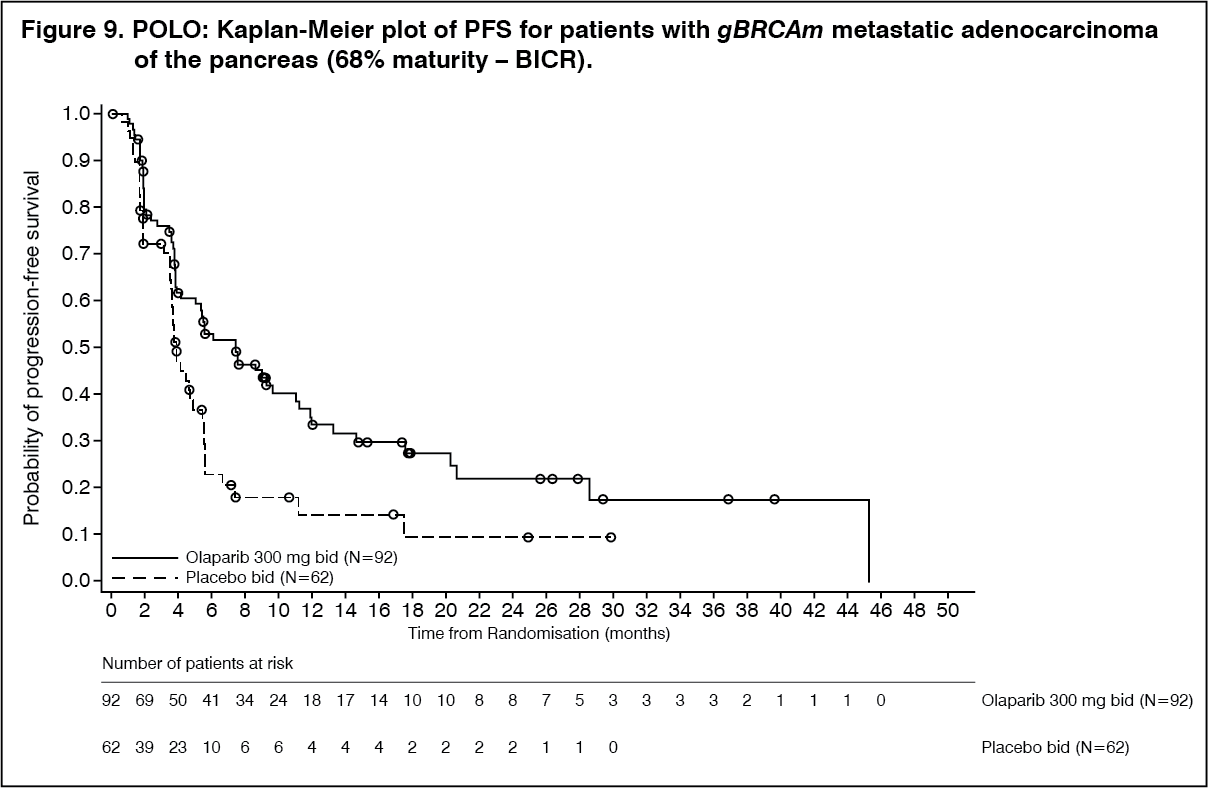 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image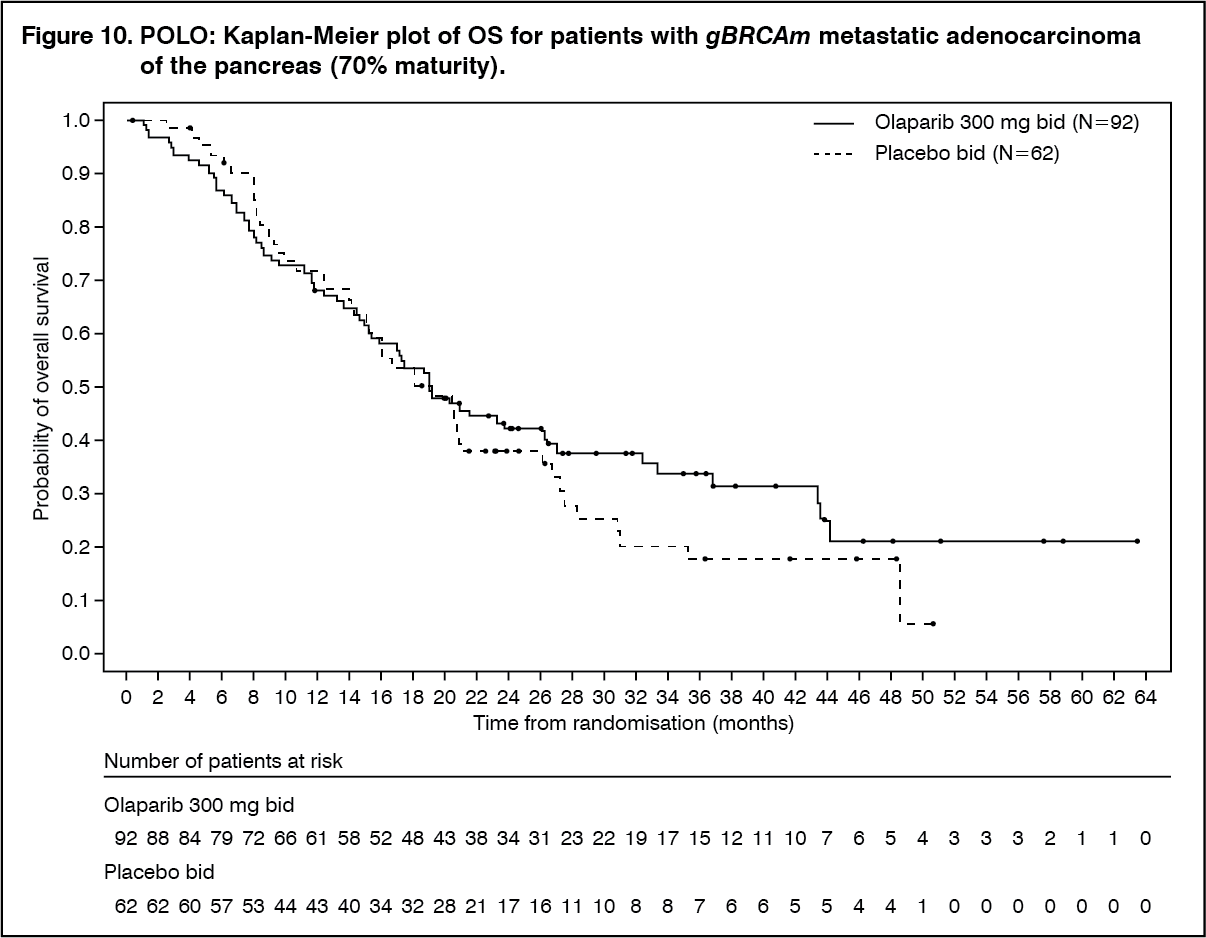 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatient-reported HRQoL was assessed using the EORTC QLQ-C30. A 10-point change was pre-defined as clinically meaningful on a 0-100 points global HRQoL scale. The adjusted mean change from baseline in global HRQoL score across all timepoints up to 6 months was -1.20 ± 1.42 in the olaparib group (n=84) and 1.27 ± 1.95 in the placebo group (n=54), with a corresponding estimated difference of -2.47 points (95% CI, -7.27 to 2.33). These results should be interpreted cautiously as this secondary endpoint was not adjusted for multiplicity testing in the statistical analysis.
Homologous recombination repair gene mutated metastatic castration-resistant prostate cancer: PROfound was a Phase III randomised, open-label, multicentre trial that evaluated the efficacy of Lynparza (300 mg [2 x 150 mg tablets] twice daily) versus a comparator arm of investigator's choice of NHA ([new hormonal agent] enzalutamide or abiraterone acetate) in men with metastatic castration-resistant prostate cancer (mCRPC). Patients needed to have progressed on prior NHA for the treatment of metastatic prostate cancer and/or CRPC and have a tumour mutation in one of 15 genes involved in the homologous recombination repair (HRR) pathway. All patients continued on a luteinising hormone releasing hormone (LHRH) analogue or had prior bilateral orchiectomy.
Patients were divided into two cohorts based on HRR gene mutation status. Patients with mutations in either BRCA1, BRCA2 or ATM were randomised in Cohort A; patients with mutations among 12 other genes involved in the HRR pathway (BARD1, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D or RAD54L) were randomised in Cohort B. Patients with co-mutations (BRCA1, BRCA2 or ATM plus a Cohort B gene) were randomised in Cohort A. Although patients with PPP2R2A gene mutations were enrolled in the trial, Lynparza has an unfavourable benefit-risk in patients with this gene mutation.
The study randomised 387 patients (2:1 randomisation: 256 olaparib and 131 comparator); in Cohort A there were 245 patients (162 olaparib and 83 comparator) and in Cohort B there were 142 patients (94 olaparib and 48 comparator). Patients were stratified by prior taxane use and evidence of measurable disease. Treatment was continued until disease progression. Patients randomised to comparator were given the option to switch to olaparib upon confirmed radiological BICR progression.
Patients with HRR gene mutations were identified based on prostate cancer tissue specimen testing by the Foundation Medicine FoundationOne HRR clinical trial assay performed in a CLIA certified laboratory (CLIA HRR Clinical Trial Assay) or from reanalysis of data from a prior Foundation Medicine test.
Demographics and baseline characteristics were generally well balanced between the olaparib and comparator arms in Cohort A+B. Median age was 69 years in both arms. Prior therapy in the olaparib arm was 66% taxane, 40% enzalutamide, 38% abiraterone acetate and 20% both enzalutamide and abiraterone acetate. Prior therapy in the comparator arm was 64% taxane, 41% enzalutamide, 41% abiraterone acetate and 18% both enzalutamide and abiraterone acetate. Fifty-eight percent (58%) of patients in the olaparib arm and 55% in the comparator arm had measurable disease at study entry. The proportion of patients with bone, distant lymph node, locoregional lymph node, liver and respiratory metastases was 85%, 39%, 21%, 10% and 17%, respectively in the olaparib arm and 86%, 39%, 24%, 14% and 12%, respectively in the comparator arm. Most patients in both treatment arms had an ECOG of 0 or 1 (95%). Baseline pain scores (BPI-SF worst pain) were 0-<2 (48%), 2-3 (12%) or >3 (36%) in the olaparib arm and 0-<2 (44%), 2-3 (10%) or >3 (43%) in the comparator arm. Median baseline PSA was 68.22 µg/L in the olaparib arm and 106.49 µg/L in the comparator arm.
The primary endpoint of the study was radiological progression free survival (rPFS) in Cohort A determined by BICR using RECIST 1.1 (soft tissue) and Prostate Cancer Working Group (PCWG3) (bone). Key secondary endpoints included confirmed objective response rate (ORR) by BICR (Cohort A), rPFS by BICR (Cohort A+B), time to pain progression (TTPP) (Cohort A) and overall survival (OS) (Cohort A).
Other secondary endpoints in Cohort A and Cohort A+B included time to first symptomatic skeletal-related event (SSRE), duration of response (DoR), time to opiate use for cancer-related pain, confirmed soft tissue ORR, prostate specific antigen (PSA50) response, circulating tumour cells (CTC) conversion rate, time to second progression or death (PFS2) and disease-related symptoms and health related quality of life [HRQoL] (pain progression, pain severity progression, pain interference and pain palliation). Other secondary end-points in Cohort B included rPFS by BICR. In addition, other secondary end-points in Cohort B and Cohort A+B included confirmed ORR by BICR, OS and time to pain progression.
The study demonstrated a clinically meaningful and statistically significant improvement in BICR assessed rPFS for olaparib vs comparator in Cohort A and also in Cohort A+B.
In Cohort A there was a statistically significant and clinically meaningful improvement in confirmed radiological ORR by BICR for patients with measurable disease at baseline in the olaparib arm vs comparator and an improvement observed in confirmed radiological ORR Cohort A+B. There was a statistically significant and clinical meaningful delay in TTPP in the olaparib arm compared with the investigators choice of NHA arm in Cohort A and the results in Cohort A+B were consistent with Cohort A.
The final analysis of OS demonstrated a statistically significant improvement in OS in patients randomised to Lynparza compared to patients in the investigators choice of NHA arm in Cohort A. (See Table 13, Figures 11, 12 and 13.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image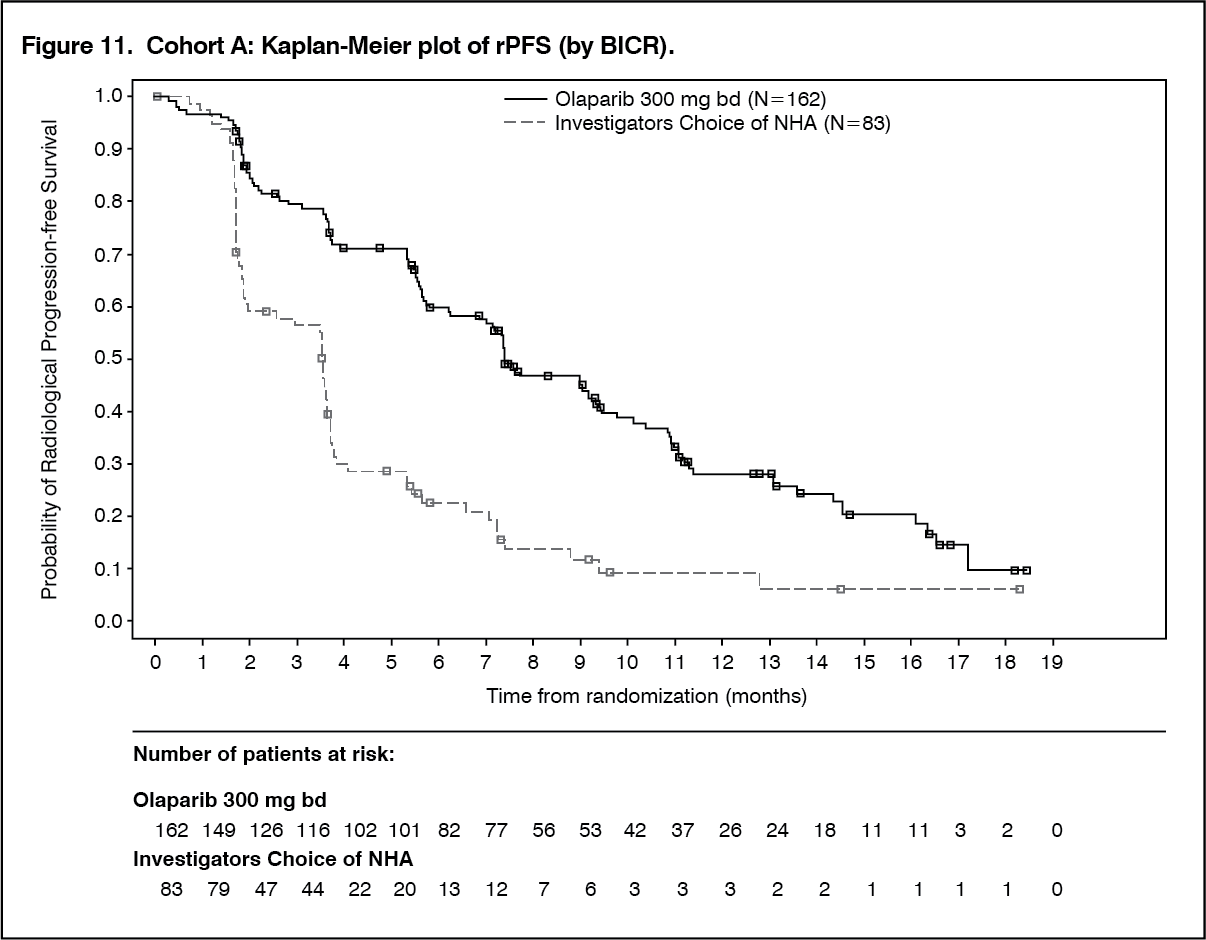 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image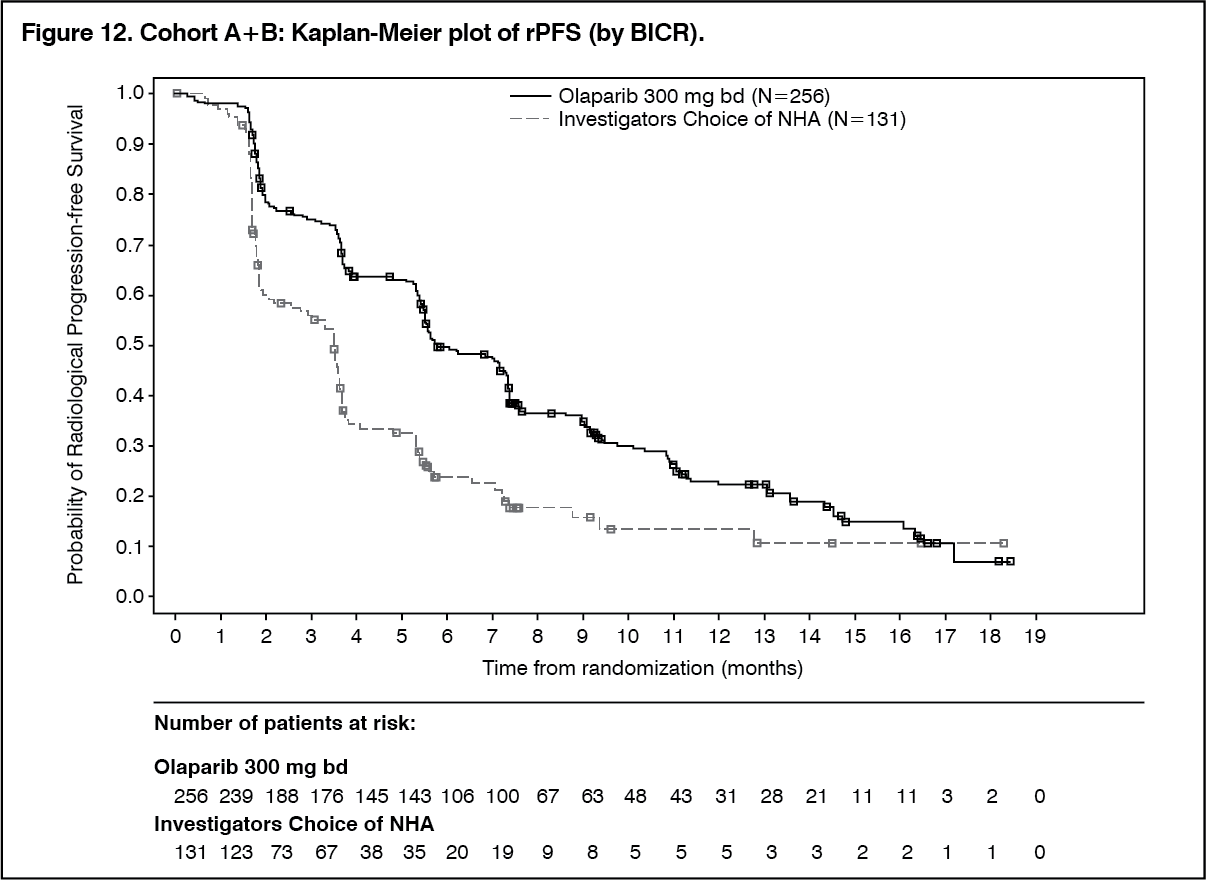 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image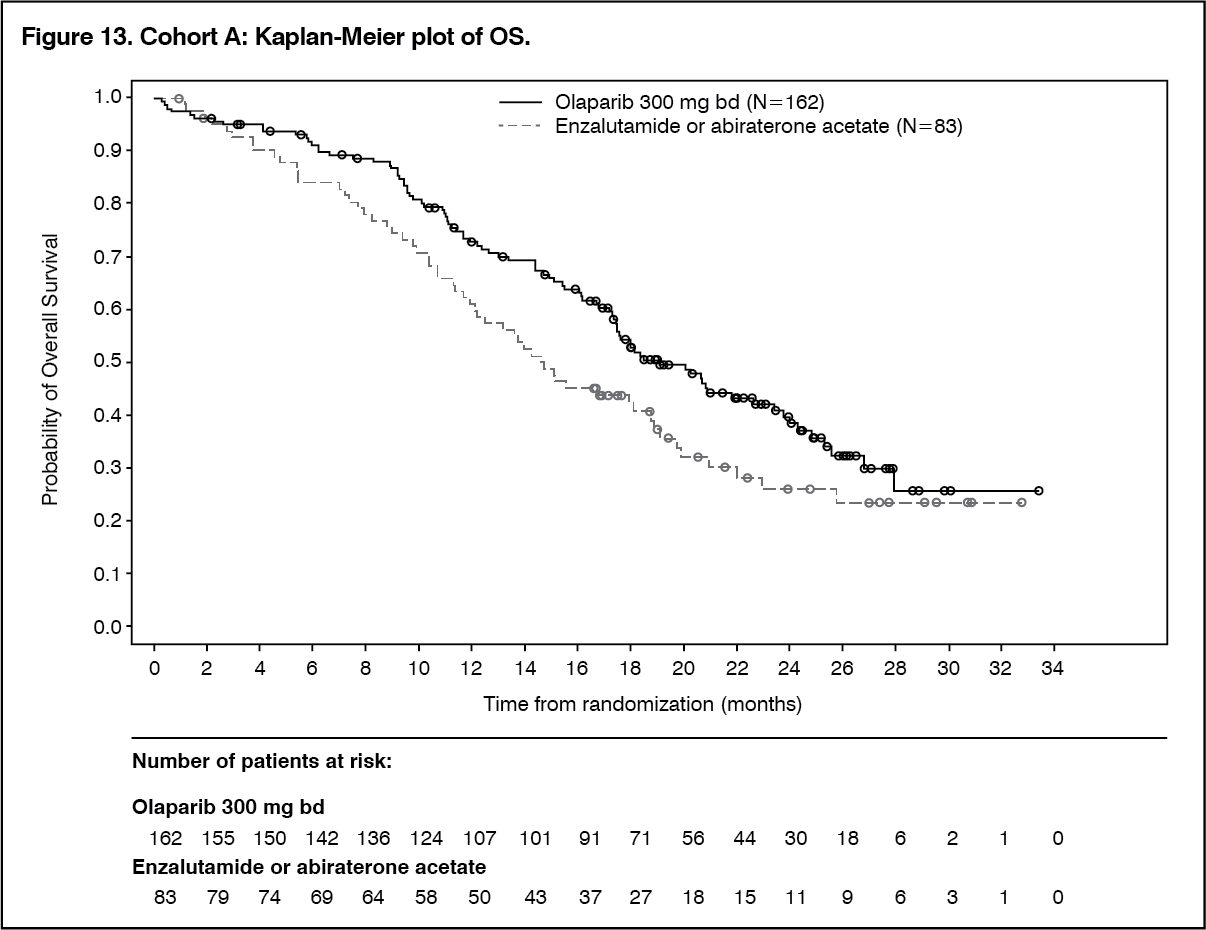 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn Cohort A and Cohort A+B the benefit of olaparib over investigators choice of NHA was maintained across all pre-defined subgroups, with clinically meaningful reductions in the risk of progression or death in olaparib-treated patients (ranging from 39% to 75% in Cohort A and from 23% to 88% in Cohort A+B).
The prevalence of the mutations in Cohort B did not provide sufficient power to test independently. In Cohort B exploratory analysis, the median rPFS was 4.8 months for olaparib vs 3.3 months for comparator with a HR of 0.88 (95% CI 0.58, 1.36; p=0.3976 [nominal]). Two patients (3.7%) in the olaparib arm and 2 patients (8.3%) in the comparator arm with measurable disease at baseline had a confirmed radiological objective response. The odds ratio (OR) and 95% CIs were not calculated due to the small number of responders. The final OS analysis had events in 100/142 patients. Median OS was 14.1 months for olaparib vs 11.5 months for comparator with a HR of 0.96 (95% CI 0.63, 1.49; p=0.7921, nominal).
Olaparib improved overall adjusted mean change (based in mixed model of repeated measures analysis) from baseline scores in HRQoL (FACT-P total score, FACT-General total score, Trial Outcome Index), functioning (physical well-being, functional well-being) and prostate cancer symptoms (PCS, FACT Advanced Prostate Symptom Index-6) compared with investigators choice of NHA during treatment demonstrating that patients in the olaparib arm experienced improvement in HRQoL, functioning and prostate cancer symptoms compared with patients in the investigators choice of NHA arm in Cohort A. The analysis of adjusted mean change from baseline in HRQoL, functioning and prostate cancer symptoms scores in Cohort A were consistent with results in Cohort A+B.
Effect on the QT interval: There is no clinically relevant effect of olaparib on cardiac repolarisation (as evaluated by an effect on the QT interval) following 300 mg twice daily multiple dosing of olaparib.
Pharmacokinetics: General: Hard capsule: The pharmacokinetics of olaparib at the 400 mg twice daily capsule dose are characterised by an apparent plasma clearance of ~8.6 L/h, an apparent volume of distribution of ~167 L and a terminal half-life of 11.9 hours.
FC tablet: The pharmacokinetics of olaparib at the 300 mg tablet dose are characterised by an apparent plasma clearance of ~7 L/h, an apparent volume of distribution of ~158 L and a terminal half-life of 15 hours. On multiple dosing, an AUC accumulation ratio of 1.8 was observed and PK appeared to be time-dependent to a small extent.
Absorption: Hard capsule: Following oral administration of olaparib via the capsule formulation, absorption is rapid with peak plasma concentrations typically achieved between 1 to 3 hours after dosing. On multiple-dosing there is no marked accumulation, with steady state exposures achieved within ~3 to 4 days.
Co-administration with food slowed the rate (Tmax delayed by 2 hours) and increased the extent of absorption of olaparib (AUC increased by approximately 20%). Consequently, patients should take Lynparza at least one hour after food, and should refrain from eating for 2 hours afterwards (see Dosage & Administration).
FC tablet: Following oral administration of olaparib via the tablet formulation (2 x 150 mg), absorption is rapid with median peak plasma concentrations typically achieved 1.5 hours after dosing.
Co-administration with food slowed the rate (tmax delayed by 2.5 hours and Cmax reduced by approximately 21%) but did not significantly affect the extent of absorption of olaparib (AUC treatment ratio: 1.08; 90% CI: 1.01, 1.16). Consequently, patients should take Lynparza without regard to food (see Dosage & Administration).
Distribution: The in vitro plasma protein binding is approximately 82% (hard capsule) at clinically relevant concentrations of 10 μg/mL; (FC tablet) at 10 μg/mL which is approximately Cmax.
In vitro, human plasma protein binding of olaparib was dose-dependent; the fraction bound was approximately 91% at 1 μg/mL, reducing to 82% at 10 μg/mL and to 70% at 40 μg/mL. In solutions of purified proteins, the olaparib fraction bound to albumin was approximately 56%, which was independent of olaparib concentrations. Using the same assay, the fraction bound to alpha-1 acid glycoprotein was 29% at 10 μg/mL with a trend of decreased binding at higher concentrations.
Metabolism: In vitro, CYP3A4/5 were shown to be the enzymes primarily responsible for the metabolism of olaparib.
Following oral dosing of 14C-olaparib to female patients, unchanged olaparib accounted for the majority of the circulating radioactivity in plasma (70%) and was the major component found in both urine and faeces (15% and 6% of the dose respectively). The metabolism of olaparib is extensive with the main site of metabolism being the piperazine and fluorobenzyl ring structures. The majority of the metabolism was attributable to oxidation reactions with a number of the components produced undergoing subsequent glucuronide or sulphate conjugation. Up to 20, 37 and 20 metabolites were detected in plasma, urine and faeces respectively, the majority of them representing <1% of the dosed material. A ring-open piperazin-3-ol moiety, and two mono-oxygenated metabolites (each ~10%) were the major circulating components, with one of the mono-oxygenated metabolites also being the major metabolite in the excreta (6% and 5% of the urinary and faecal radioactivity respectively).
In vitro, olaparib produced little/no inhibition of UGT1A4, UGT1A9, UGT2B7, or CYPs 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 and is not expected to be a clinically significant time dependent inhibitor of any of these CYP enzymes. Olaparib inhibited UGT1A1 in vitro, however, PBPK simulations suggest this is not of clinical importance. Based on evaluation using enzyme activity, olaparib was not an inducer of CYP2C9 or 2C19. In vitro, olaparib is a substrate of and inhibits the efflux transporter P-gp (IC50=76 μM), however, this is unlikely to be of clinical significance.
In vitro, data also show that olaparib is not a substrate for OATP1B1, OATP1B3, OCT1, BCRP or MRP2, is a weak inhibitor of BCRP and not an inhibitor of OATP1B3, OAT1 or MRP2.
Excretion: Following a single dose of 14C-olaparib, ~86% of the dosed radioactivity was recovered within a 7-day collection period, ~44% via the urine and ~42% via the faeces. The majority of the material was excreted as metabolites.
Special populations: In population based PK analyses, patient age, gender (FC tablet), bodyweight, tumour location (FC tablet) or race (including White and Japanese patients) were not significant covariates.
Effect of Renal Impairment: Following a single oral 300 mg dose of olaparib (tablet formulation) to patients with mild renal impairment (creatinine clearance: 51 to 80 mL/min), AUC increased by 24% and Cmax by 15% compared with patients with normal renal function. No Lynparza dose adjustment is required for patients with mild renal impairment.
Following a single oral 300 mg dose of olaparib (tablet formulation) to patients with moderate renal impairment (creatinine clearance: 31 to 50 mL/min), AUC increased by 44% and Cmax by 26% compared with patients with normal renal function. Lynparza dose adjustment is recommended for patients with moderate renal impairment (see Dosage & Administration).
Olaparib has not been studied in patients with severe renal impairment or end-stage renal disease (creatinine clearance ≤30 ml/min).
Effect of Hepatic Impairment: Following a single oral 300 mg dose of olaparib (tablet formulation) to patients with mild hepatic impairment (Child-Pugh classification A) AUC increased by 15% and Cmax by 13% and to patients with moderate hepatic impairment (Child-Pugh classification B) AUC increased by 8% and Cmax decreased by 13% compared with patients with normal hepatic function. No Lynparza dose adjustment is required in patients with mild or moderate hepatic impairment (see Dosage & Administration).
Olaparib has not been studied in patients with severe hepatic impairment (Child-Pugh classification C).
Toxicology: Preclinical safety data: Mutagenicity: Olaparib showed no mutagenic potential, but was clastogenic in mammalian cells in vitro. When dosed orally to rats, olaparib induced micronuclei in bone marrow. This clastogenicity is consistent with the primary pharmacology of olaparib and indicates potential for genotoxicity in man.
Repeat-dose toxicity: In repeat-dose toxicity studies of up to 6 months duration in rats and dogs, daily oral doses of olaparib were well-tolerated. The major primary target organ for toxicity in both species was the bone marrow, with associated changes in peripheral haematology parameters. These findings occurred at exposures below those seen clinically and were largely reversible within 4 weeks of cessation of dosing. Studies using human bone marrow cells also showed that direct exposure to olaparib can result in toxicity to bone marrow cells in ex vivo assays.
Reproductive toxicology: Olaparib had no effect on fertility in male rats. In a female fertility study in rats, extended oestrus was observed in some animals although mating performance and fertility was not affected. Embryofoetal survival was reduced in this study.
In rat embryofoetal development studies, olaparib caused reduced embryofoetal survival, reduced foetal weight and foetal developmental abnormalities (including visceral and skeletal abnormalities, and major eye and vertebral/rib malformations) at dose levels that did not induce significant maternal toxicity.
Carcinogenicity: Carcinogenicity studies have not been conducted with olaparib.