Lamotrigine tablets should be swallowed whole and should not be chewed or crushed.
If a calculated dose of Lamotrix, e.g. for use in children (epilepsy only) or patients with hepatic impairment, cannot be divided into multiple lower strength tablets, the dose to be administered is that equal to the nearest lower strength of whole tablets.
It is strongly recommended that therapy with lamotrigine is initiated at the recommended doses. Careful incremental titration of the dose may decrease the severity of skin rashes. There are suggestions, yet to be proven, that the risk of severe, potentially life-threatening rash may be increased by co-administration of lamotrigine with valproate.
However, cases have been reported in the absence of these factors. Therefore, it is important that the dosing recommendations to be followed closely.
Restarting Therapy: Prescribers should assess the need for escalation to maintenance dose when restarting lamotrigine in patients who have discontinued lamotrigine for any reason, since the risk of serious rash is associated with high initial doses and exceeding the recommended dose escalation for lamotrigine (see Precautions). The greater the interval of time since the previous dose, the more consideration should be given to escalation to the maintenance dose. When the interval since discontinuing lamotrigine exceeds five half-lives (see Pharmacology: Pharmacokinetics under Actions), lamotrigine should generally be escalated to the maintenance dose according to the appropriate schedule.
It is recommended that lamotrigine not be restarted in patients who have discontinued due to rash associated with prior treatment with lamotrigine unless the potential benefit clearly outweighs the risk.
Epilepsy
: When concomitant antiepileptic drugs are withdrawn to achieve lamotrigine monotherapy or other AEDs are added on to treatment regimes containing lamotrigine, consideration should be given to the effect this may have on lamotrigine pharmacokinetics (see Interactions).
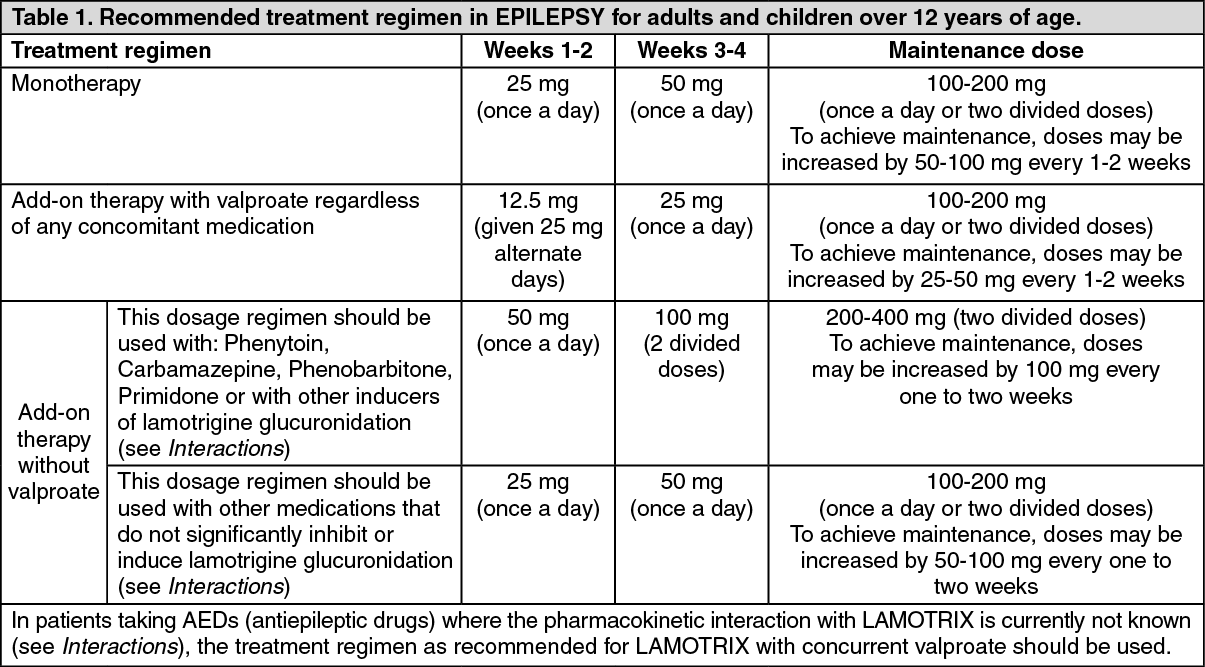
Adults and children over 12 years of age (see Table 1): Dosage in epilepsy monotherapy: The initial LAMOTRIX dose in monotherapy is 25mg once a day for two weeks, followed by 50mg once a day for two weeks. Thereafter, the dose should be increased by a maximum of 50mg-100mg every 1-2 weeks until the optimal response is achieved. The usual maintenance dose to achieve optimal response is 100 - 200 mg/day given once a day or as two divided doses. Some patients have required 500mg/day of lamotrigine to achieve the desired response.
Because of a risk of rash the initial dose and the subsequent dose escalation should not be exceeded (see Precautions).
Dosage in epilepsy add-on therapy: In patients taking valproate with / without any other anti-epileptic drug (AED) the initial LAMOTRIX dose is 25mg every alternate day for two weeks, followed by 25mg once a day for two weeks. Thereafter, the dose should be increased by a maximum of 25-50mg every 1 - 2 weeks until the optimal response is achieved. The usual maintenance dose to achieve optimal response is 100 - 200mg/day given once a day or in two divided doses. In those patients taking concomitant AEDs or other medications (see Interactions) that induce lamotrigine glucuronidation with/without other AEDs (except valproate), the initial LAMOTRIX dose is 50 mg once a day for two weeks, followed by 100 mg/day given in two divided doses for two weeks. Thereafter, the dose should be
increased by a maximum of 100mg every 1-2 weeks until the optimal response is achieved. The usual maintenance dose to achieve optimal response is 200-400 mg/day given in two divided doses. Some patients have required 700 mg/day of lamotrigine to achieve the desired response.
In those patients taking other medications that do not significantly inhibit or induce lamotrigine glucuronidation (see Interactions), the initial Lamotrix dose is 25 mg once a day for two weeks, followed by 50 mg once a day for two weeks. Thereafter, the dose should be increased by a maximum of 50 to 100 mg every one to two weeks until the optimal response is achieved. The usual maintenance dose to achieve an optimal response is 100 to 200 mg/day given once a day or as two divided doses. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Because of a risk of rash the initial dose and subsequent dose escalation should not be exceeded (see Precautions).
Children aged 2 to 12 years: In patients taking valproate with/without any other anti-epileptic drug (AED), the initial Lamotrix dose is 0.15mg/kg bodyweight/day given once a day for two weeks, followed by 0.3mg/kg/day given once a day for two weeks. Thereafter, the dose should be increased by a maximum of 0.3mg/kg every 1-2 weeks until the optimal response is achieved. The usual maintenance dose to achieve optimal response is 1-5 mg/kg/day given once a day or in two divided doses, with a maximum of 200 mg/day.
In those patients taking concomitant AEDs or other medications (see Interactions) that induce lamotrigine glucuronidation with/without other AEDs (except valproate) the initial lamotrigine dose is 0.6 mg/kg bodyweight/day given in two divided doses for two weeks, followed by 1.2 mg/kg/day given in two divided doses for two weeks. Thereafter, the dose should be increased by a maximum of 1.2mg/kg every 1-2 weeks until the optimal response is achieved. The usual maintenance dose to achieve optimal response is 5-15 mg/kg/day given in two divided doses, with a maximum of 400 mg/day.
To ensure a therapeutic dose is maintained the weight of a child must be monitored and the dose reviewed as weight changes occur. (See Table 2.)
Click on icon to see table/diagram/image
Because of the risk of rash the initial dose and subsequent dose escalation should not be exceeded (see Precautions).
It is likely that patients aged 2-6 years will require a maintenance dose at the higher end of the recommended range.
Children aged less than two years: Lamotrigine has not been studied as monotherapy in children less than 2 years of age or as add-on therapy in children less than 1 month of age. The safety and efficacy of lamotrigine as add on therapy of partial seizures in children aged 1 month to 2 years has not been established (see Pharmacology: Pharmacodynamics: Clinical Studies under Actions). Therefore LAMOTRIX is not recommended in children less than 2 years of age.
Bipolar Disorder: Adults (18 years of age and over): Because of the risk of rash the initial dose and subsequent dose escalation should not be exceeded (see Precautions).
LAMOTRIX is recommended for use in bipolar patients at risk for a future depressive episode. The following transition regimen should be followed to prevent recurrence of depressive episodes. The transition regimen involves escalating the dose of LAMOTRIX to a maintenance stabilization dose over six weeks (see Table 3) after which other psychotropic and/or anti-epileptic drugs can be withdrawn, if clinically indicated (see Table 4).
Adjunctive therapy should be considered for the prevention of manic episodes, as efficacy with lamotrigne in mania has not been conclusively established. There is no evidence of an increased risk of mania, hypomania or mixed type episodes with lamotrigine treatment compared to placebo. (See Table 3.)
Click on icon to see table/diagram/image
Adjunct therapy with inhibitors of lamotrigine glucuronidation e.g. valproate: In patients taking glucuronidation inhibiting concomitant drugs such as valproate the initial LAMOTRIX dose is 25mg every alternate day for two weeks, followed by 25mg once a day for two weeks. The dose should be increased to 50mg once a day (or in two divided doses) in week 5. The usual target dose to achieve optimal response is 100mg/day given once a day or in two divided doses. However, the dose can be increased to a maximum daily dose of 200mg, depending on clinical response.
Adjunct therapy with inducers of lamotrigine glucuronidation in patients NOT taking inhibitors such as Valproate. This dosage regimen should be used with: Phenytoin, Carbamazepine, Phenobarbitone, Primidone or with others inducers of lamotrigine glucuronidation (see Interactions).
In those patients currently taking drugs that induce lamotrigine glucuronidation and not taking Valproate, the initial LAMOTRIX dose is 50mg once a day for two weeks, followed by 100mg/day given in two divided doses for two weeks. The dose should be increased to 200mg/day given as two divided doses in week 5. The dose may be increased in week 6 to 300mg/day however, the usual target dose to achieve optimal response is 400mg/day given in two divided doses which may be given from week 7.
Monotherapy with LAMOTRIX or Adjunctive therapy in patients taking other medications that do not significantly inhibit or induce lamotrigine glucuronidation (see Interactions): The initial Lamotrix dose is 25mg once a day for two weeks, followed by 50mg once a day (or in two divided doses) for two weeks. The dose should be increased to 100mg/day in week 5. The usual target dose to achieve optimal response is 200mg/day given once a day or as two divided doses. However, a range of 100 to 400mg was used in clinical trials.
Once the target daily maintenance stabilization dose has been achieved, other psychotropic medications may be withdrawn as laid out in the dosage schedule as follows (see Table 4).
Click on icon to see table/diagram/image
Following withdrawal of adjunct therapy with inhibitors of lamotrigine glucuronidation e.g. Valproate: The dose of Lamotrix should be increased to double the original target stabilisation dose and maintained at this, once Valproate has been terminated.
Following withdrawal of adjunct therapy with inducers of lamotrigine glucuronidation depending on original maintenance dose. This dosage regimen should be used with: Phenytoin, Carbamazepine, Phenobarbitone, Primidone or with others drugs known to induce of lamotrigine glucuronidation (see Interactions). The dose of LAMOTRIX should be gradually reduced over three weeks as the glucuronidation inducer is withdrawn.
Following withdrawal of adjunct therapy with medications that do not significantly inhibit or induce lamotrigine glucuronidation (see Interactions): The target dose achieved in the dose escalation programme should be maintained throughout withdrawal of the other medication.
Adjustment of LAMOTRIX daily dosing in patients with BIPOLAR DISORDER following addition of other medications: There is no clinical experience in adjusting the lamotrigine daily dose following the addition of other medications.
However, based on drug interaction studies, the following recommendations can be made (see Table 5, as follows): (See Table 5.)
Click on icon to see table/diagram/image
Discontinuation of LAMOTRIX in adult patients with BIPOLAR DISORDER: In clinical trials, there was no increase in the incidence, severity or type of adverse experiences, following abrupt termination of lamotrigine versus placebo. Therefore patients may terminate LAMOTRIX without a step-wise reduction of dose.
Children and adolescents (less than 18 years of age): LAMOTRIX is not indicated for use in bipolar disorder in children and adolescents aged less than 18 years old (see Precautions). Safety and efficacy of lamotrigine in bipolar disorder has not been evaluated in this age group. Therefore, a dosage recommendation cannot be made.
General dosing recommendations for lamotrix in special patient populations: Women taking hormonal contraceptives:
Starting LAMOTRIX in patients already taking hormonal contraceptives: Although an oral contraceptive has been shown to increase the clearance of lamotrigine (see Precautions and Interactions), no adjustments to the recommended dose escalation guidelines for LAMOTRIX should be necessary solely based on the use of hormonal contraceptives. Dose escalation should follow the recommended guidelines based on whether lamotrigine is added to an inhibitor of lamotrigine glucuronidation e.g. valproate; whether Lamotrix is added to an inducer of lamotrigine glucuronidation e.g. carbamazepine, Phenytoin, phenobarbital, primidone, rifampin or lopinavir/ritonavir; or whether LAMOTRIX is added in the absence of valproate, carbamazepine, phenytoin, phenobarbital, primidone, rifampicin or lopinavir/ritonavir (see Table 1 for epilepsy and Table 3 for bipolar disorder patients).
Starting hormonal contraceptives in patients already taking maintenance doses of lamotrigine and NOT taking inducers of lamotrigine glucuronidation:The maintenance dose of lamotrigine may need to be increased as much as two-fold (see Precautions & Interactions). It is recommended that from the time that the hormonal contraceptive is started, the lamotrigine dose is increased by 50 to 100 mg/day every week, according to the individual clinical response. Dose increases should not exceed this rate, unless the clinical response supports larger increases.
Stopping hormonal contraceptives in patients already taking maintenance doses of LAMOTRIX and NOT taking inducers of lamotrigine glucuronidation: The maintenance dose of lamotrigine will in most cases need to be decreased as much as 50% (see Precautions & Interactions). It is recommended to gradually decrease the daily dose of lamotrigine by 50 to 100 mg each week (at a rate not exceeding 25% of the total daily dose per week) over a period of 3 weeks, unless the clinical response indicates otherwise.
Elderly (over 65 years of age): No dosage adjustment from recommended schedule is required. The pharmacokinetics of lamotrigine in this age group do not differ significantly from a non-elderly adult population. As older patients are more likely to suffer from intercurrent illness and require medications to treat other medical conditions, lamotrigine should be used cautiously in these patients and they should be monitored regularly.
Hepatic impairment: Initial, escalation and maintenance doses should generally be reduced by approximately 50% in patients with moderate (Child-Pugh grade B) and 75% in severe (Child-Pugh grade C) hepatic impairment.
Escalation and maintenance doses should be adjusted according to clinical response (see Pharmacology: Pharmacokinetics under Actions).
Renal impairment: Caution should be exercised when administering LAMOTRIX to patients with renal failure. For patients with end-stage renal failure, initial doses of LAMOTRIX should be based on patients' AEDs regimen; reduced maintenance doses may be effective for patients with significant renal functional impairment (see Precautions). For more detailed pharmacokinetic information see Pharmacology: Pharmacokinetics under Actions.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image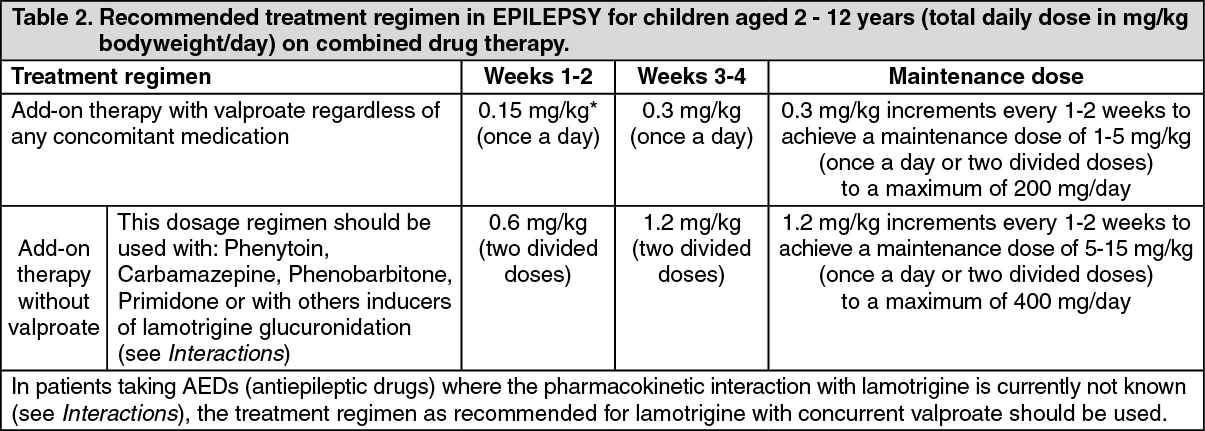 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image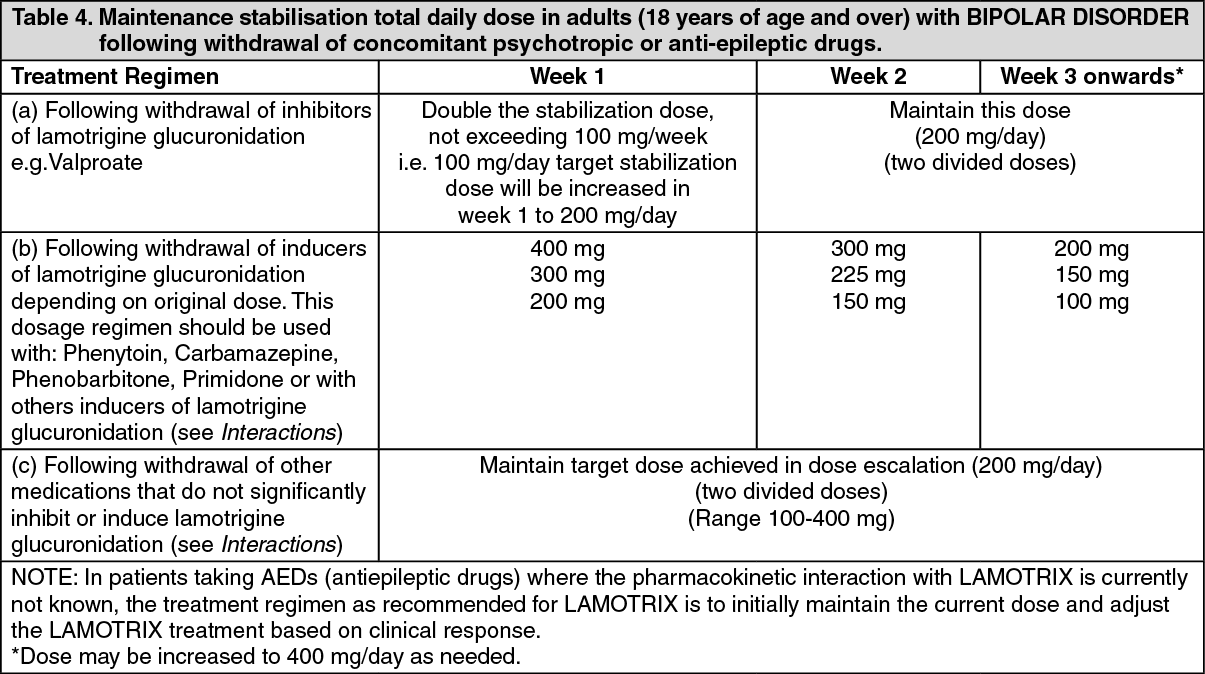 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image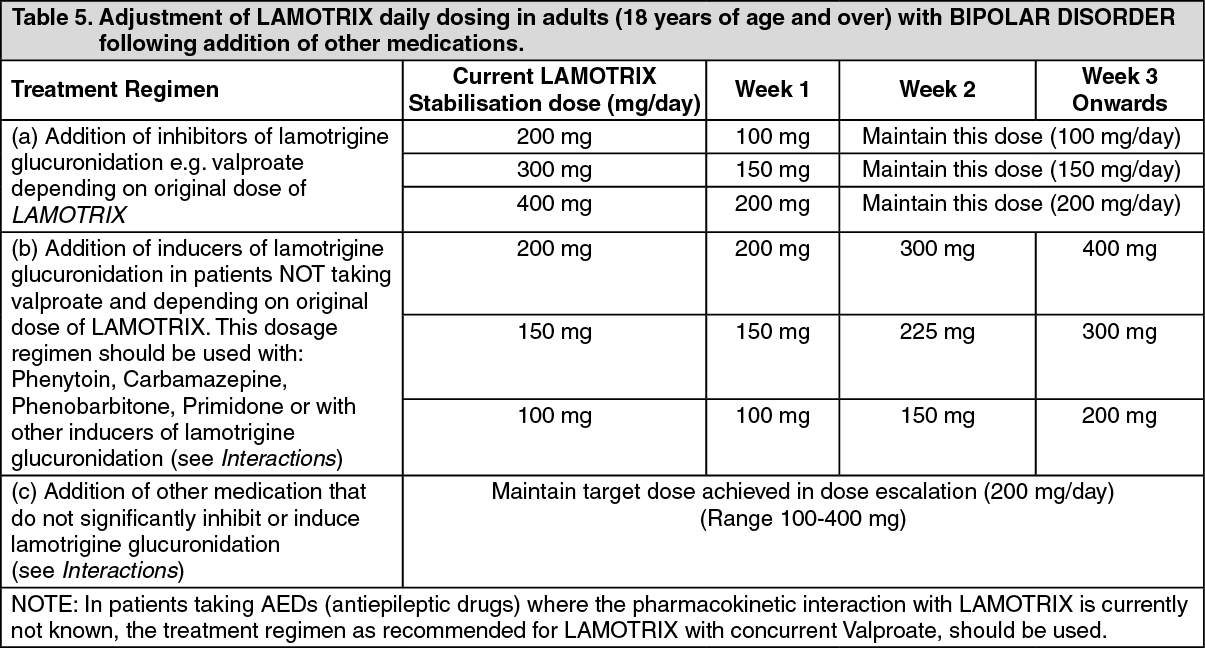 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
