Pharmacotherapeutic Group: Other antineoplastic agents.
ATC Code: L01XX41.
Pharmacology: Pharmacodynamics: HALAVEN (Eribulin mesylate) is a tubulin-targeted, non-taxane microtubule dynamics inhibitor that is a structurally simplified, synthetic analogue of the marine natural product halichondrin B.
Mechanism of action: Eribulin mesylate is a tubulin-targeted antimitotic agent that exerts its antiproliferative effects on cancer cells via a mechanistically novel mode of inhibition of microtubule dynamics. Such inhibition leads to G
2/M (Gap 2/mitosis stages of cell cycle) cell cycle blocks, disruption of mitotic spindles, and ultimately apoptotic cell death after prolonged mitotic blockage.
In addition, eribulin treatment of human breast cancer cells caused changes in morphology and gene expression as well as decreased migration and invasiveness in vitro. In mouse xenograft models of human breast cancer, eribulin treatment was associated with increased vascular perfusion and permeability in the tumor cores, resulting in reduced tumor hypoxia, and changes in the expression of genes in tumor specimens associated with a change in phenotype.
Clinical experience: Breast cancer: The efficacy of HALAVEN in breast cancer is supported by two single-arm Phase 2 studies in 403 patients and the randomized Phase 3 comparative study in 762 patients. The patients in the pivotal study had locally recurrent or metastatic breast cancer, and had previously received at least two and a maximum of five chemotherapy regimens, including an anthracycline and a taxane (unless contraindicated).
In the pivotal Phase 3 EMBRACE study, patients must have progressed within 6 months of their last therapeutic regimen. They were randomized 2:1 to receive either HALAVEN (1.4 mg/m
2 on Days 1 and 8 in a 21-day cycle administered intravenously over 2 to 5 minutes), or treatment of physician's choice (TPC), defined as any single-agent chemotherapy, hormonal treatment, or biologic therapy approved for the treatment of cancer; or palliative treatment or radiotherapy, reflecting local practice.
The study met its primary endpoint with an overall survival result that was statistically significantly better in the eribulin group compared to TPC at 55% of events. The median survival of the HALAVEN group (median: 399 days/13.1 months) compared with the TPC group (median: 324 days/10.6 months) improved by 75 days/2.5 months (HR 0.809, 95% CI: 0.660, 0.991, p=0.041).
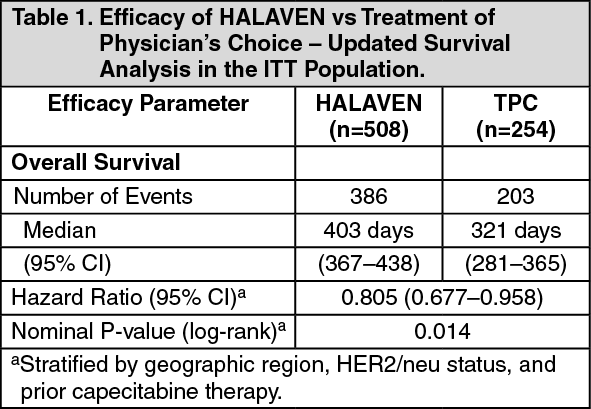
This result was confirmed with an updated overall survival analysis carried out at 77% of events with the median survival of the HALAVEN group (median: 403 days/13.2 months) compared with the TPC group (median: 321 days/10.5 months) improved by 82 days/2.7 months (HR 0.805, 95% CI: 0.677, 0.958, nominal p=0.014). (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
At the time of the original data cut-off, the PFS and ORR results according to independent and investigator's assessment are described in the following tables: (See Tables 2 and 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Liposarcoma: In liposarcoma the efficacy of eribulin is supported by the pivotal Phase 3 sarcoma study (Study 309). The patients in this study (n=452) had locally recurrent, inoperable and/or metastatic soft tissue sarcoma of one of two subtypes - leiomyosarcoma or liposarcoma. Patients had received at least two prior chemotherapy regimens, one of which must have been an anthracycline (unless contraindicated).
Patients must have progressed within 6 months of their last chemotherapeutic regimen. They were randomized 1:1 to receive either eribulin 1.23 mg/m
2 on days 1 and 8 of a 21 day cycle or dacarbazine 850 mg/m
2, 1000 mg/m
2 or 1200 mg/m
2 (dose determined by the investigator prior to randomization), every 21 days.
In Study 309, a statistically significant improvement in OS was observed in patients randomized to the eribulin arm compared to the control arm. This translated into a 2 month improvement in median OS (13.5 months for eribulin treated patients vs. 11.5 months for dacarbazine treated patients). There was no significant difference in progression-free survival or overall response rate between the treatment arms in the overall population.
Treatment effects of eribulin were limited to patients with liposarcoma (45% dedifferentiated, 37% myxoid/round cell and 18% pleomorphic in Study 309) based on pre-planned subgroup analyses of OS and PFS. There was no difference in efficacy between eribulin and dacarbazine in patients with advanced or metastatic leiomyosarcoma. (See Table 4 and Figures 2 and 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacokinetics: Distribution: The pharmacokinetics of eribulin are characterized by a rapid distribution phase followed by a prolonged elimination phase, with a mean terminal half-life of approximately 40 hr. It has a large volume of distribution (range of means 43 to 114 L/m
2) and low clearance (range of means 1.16 to 2.42 L/hr/m
2).
Eribulin is weakly bound to plasma proteins. The plasma protein binding of eribulin (100-1000 ng/mL) ranged from 49% to 65% in human plasma.
Biotransformation: Cytochrome P450 3A4 (CYP3A4) is the major enzyme responsible for the human hepatic metabolism of eribulin. Eribulin (0.05 to 5μM) did not show induction potential for CYP1A and CYP3A in human primary hepatocytes. No significant inhibition of CYP1A2, CYP2C9, CYP2C19, CYP2D6, or CYP2E1 was detected with eribulin at concentrations up to 5 μM in pooled human liver microsomes. Therefore, at the current recommended human dose eribulin is unlikely to affect plasma levels of drugs that are substrates of CYP enzymes.
Unchanged eribulin was the major circulating species in plasma following administration of
14C-eribulin to patients. Metabolite concentrations represented <0.6% of parent compound, confirming that there are no major human metabolites of eribulin.
Elimination: Eribulin is eliminated primarily in faeces. After administration of
14C-eribulin to patients, approximately 82% of the dose was eliminated in faeces and 9% in urine indicating that renal clearance is not a significant route of eribulin elimination. Based on the population PK analysis, renal impairment is not expected to significantly influence eribulin exposure.
Unchanged eribulin represented most of the total radioactivity in faeces and urine.
Hepatic impairment: A study evaluated the PK of eribulin in patients with mild (Child-Pugh A; n=7) and moderate (Child-Pugh B; n=4) hepatic impairment. Compared to patients with normal hepatic function (n=6), eribulin exposure increased 1.8-fold and 2.5-fold in patients with mild and moderate hepatic impairment, respectively. Administration of HALAVEN at a dose of 1.1 mg/m
2 to patients with mild hepatic impairment and 0.7 mg/m
2 to patients with moderate hepatic impairment resulted in similar exposure to eribulin as a dose of 1.4 mg/m
2 to patients with normal hepatic function. HALAVEN was not studied in patients with severe hepatic impairment (Child-Pugh C).
Renal impairment: Increased eribulin exposure was seen in some patients with moderately or severely impaired renal function, with high between-subject variability. The pharmacokinetics of eribulin were evaluated in a Phase I study in patients with normal renal function (Creatinine clearance: ≥ 80 ml/min; n=6), moderate (30-50 ml/min; n=7) or severe (15-<30 ml/min; n=6) renal impairment. Creatinine clearance was estimated with the Cockcroft-Gault formula. A 1.5-fold (90% Cl: 0.9-2.5) higher dose-normalised AUC
(0-inf) was observed in patients with moderate and severe renal impairment. See Dosage & Administration for treatment recommendations.
Toxicology: Preclinical safety data: The nonclinical safety of eribulin mesylate has been evaluated in the following studies: safety pharmacology, repeated toxicity including chronic toxicity, genotoxicity, and reproductive toxicity studies. No significant adverse effects were observed in any safety pharmacology studies except for transient decreases in blood pressure and heart rate in conscious dogs. Results from the toxicology studies showed that the target organ of toxicity is limited to the bone marrow, skeletal muscle, peripheral nerves, gastrointestinal system, and testes. Most of these observed toxic effects were partially or completely reversible by 2 to 4 weeks of recovery. In addition, eribulin mesylate, like other cytotoxic agents, is genotoxic and teratogenic.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image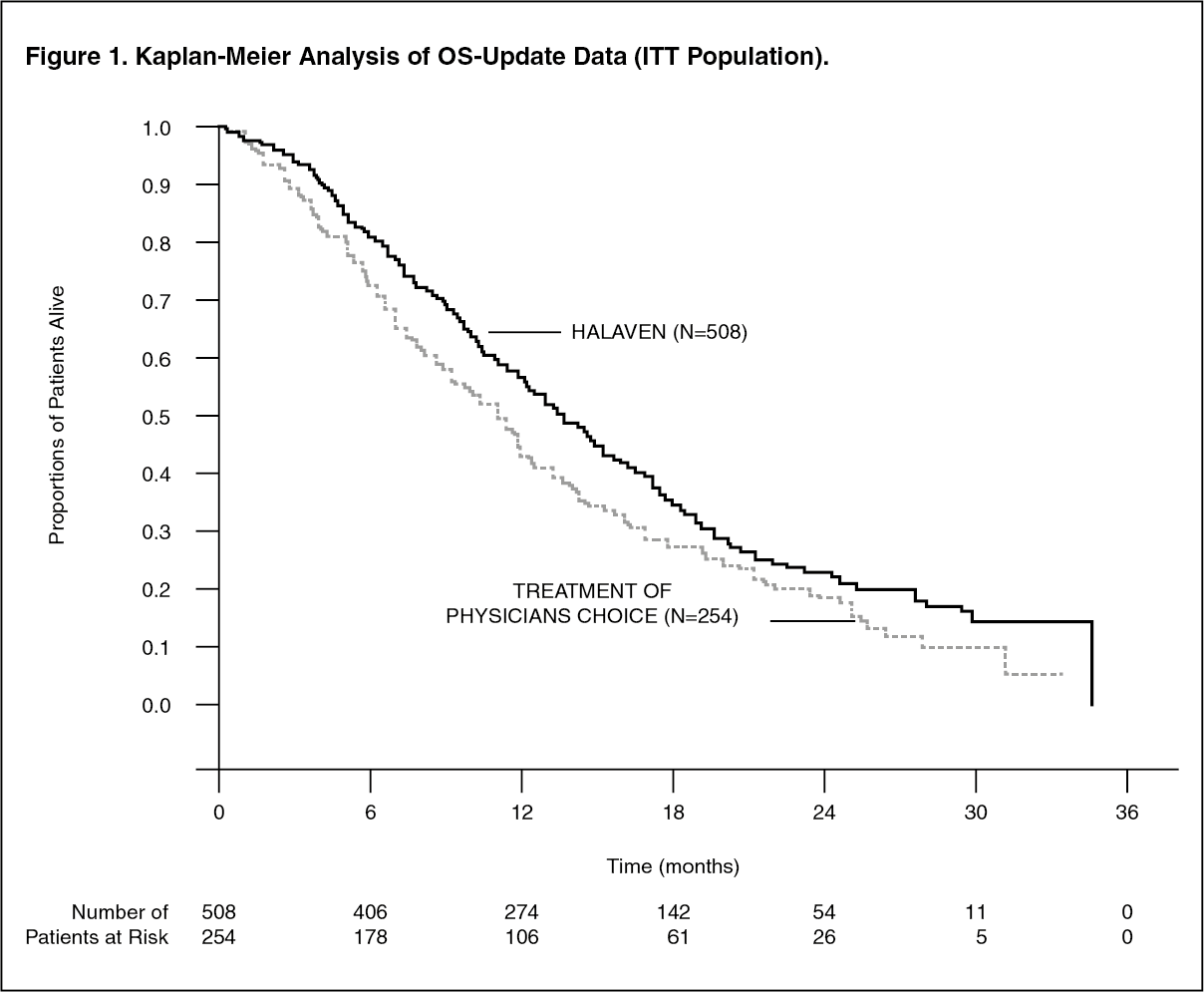 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image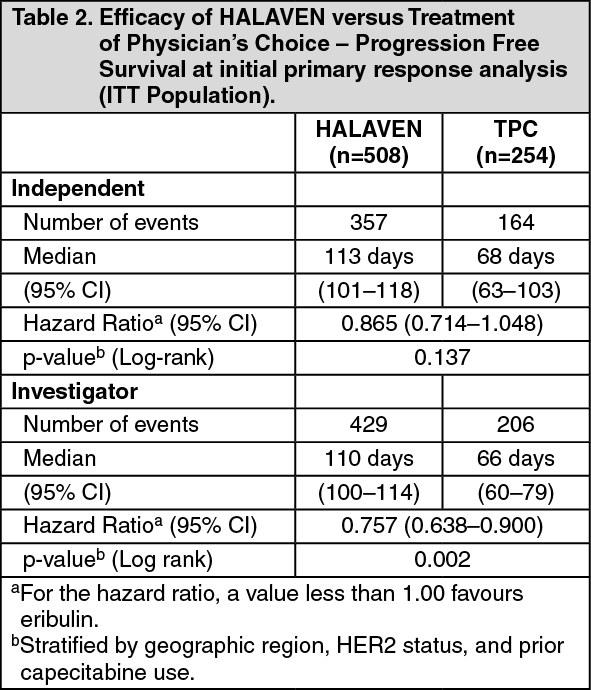 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image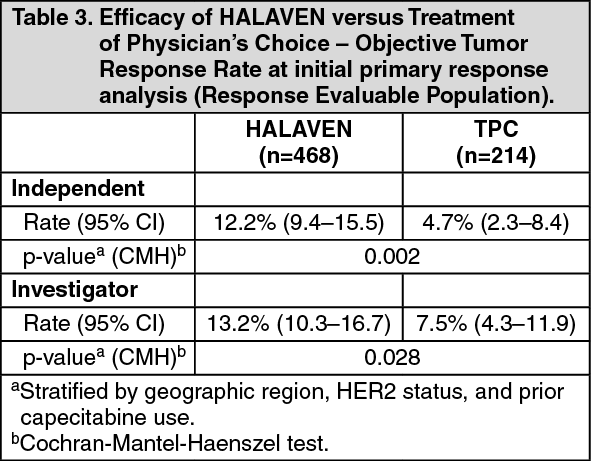 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image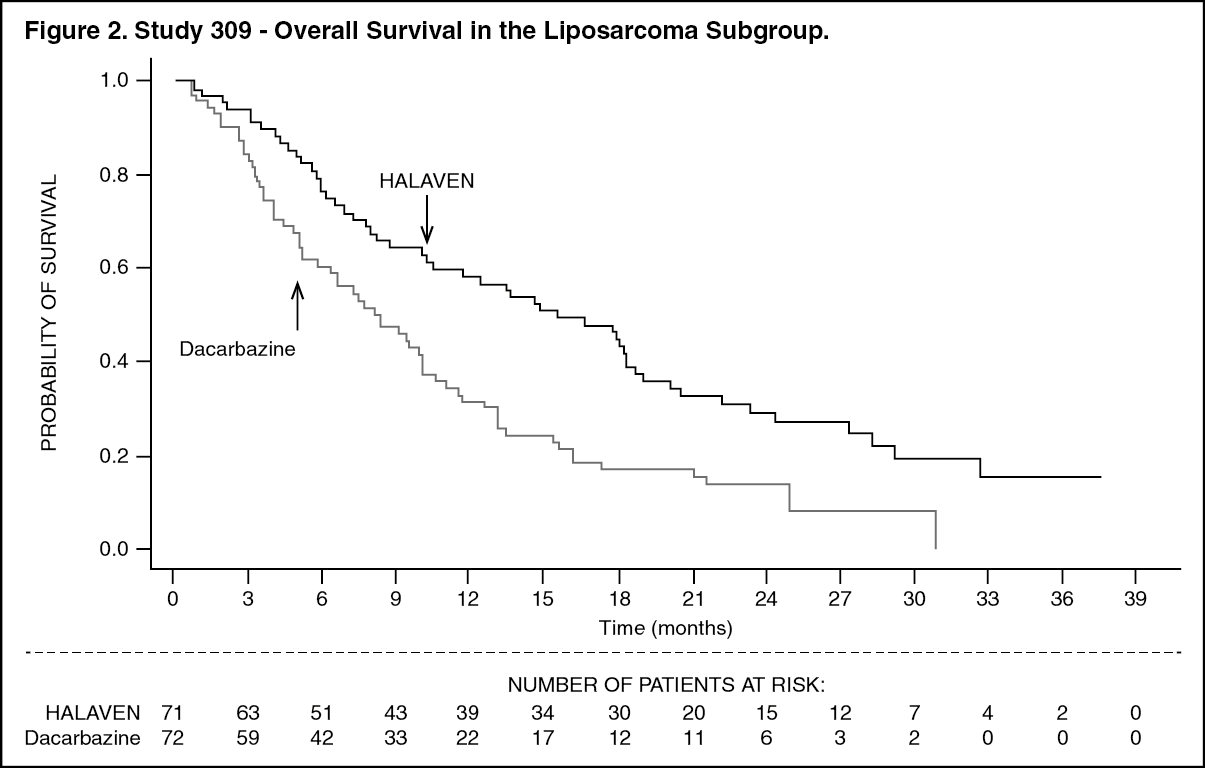 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image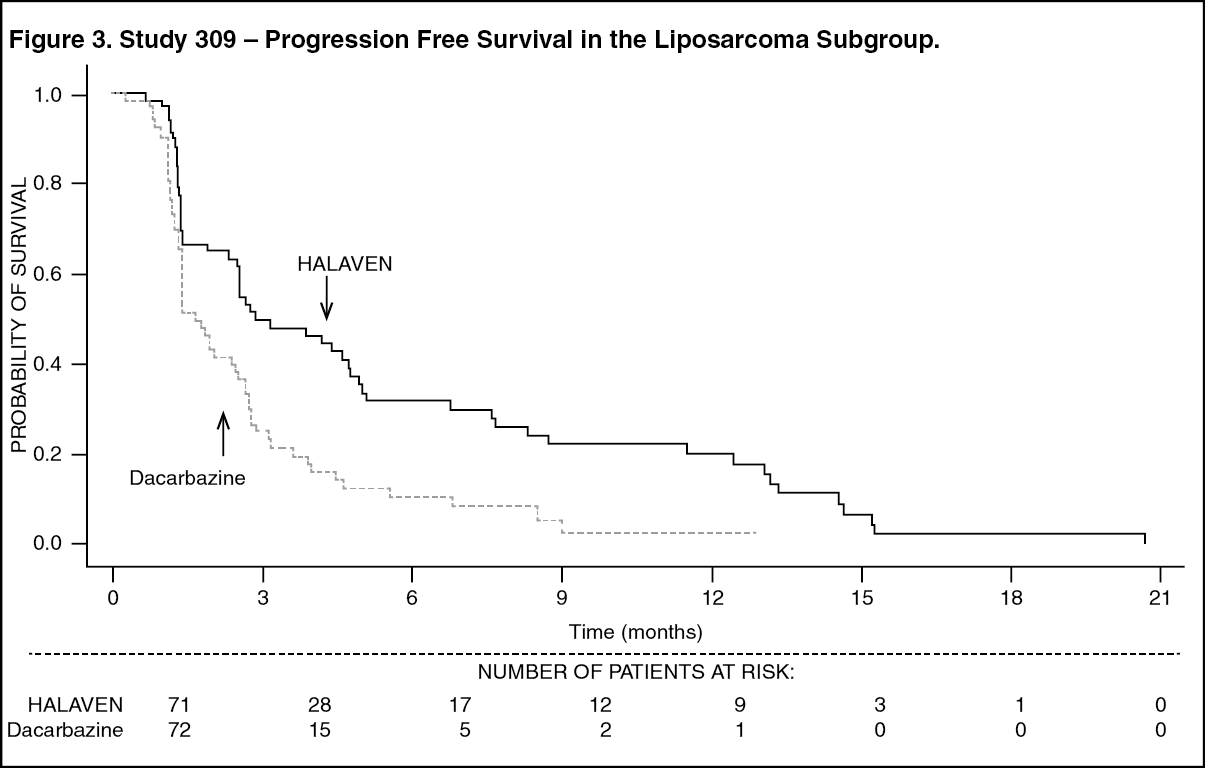 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image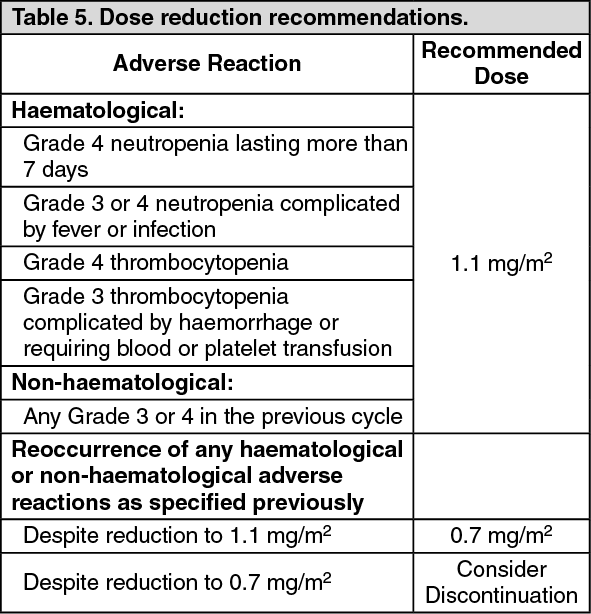 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image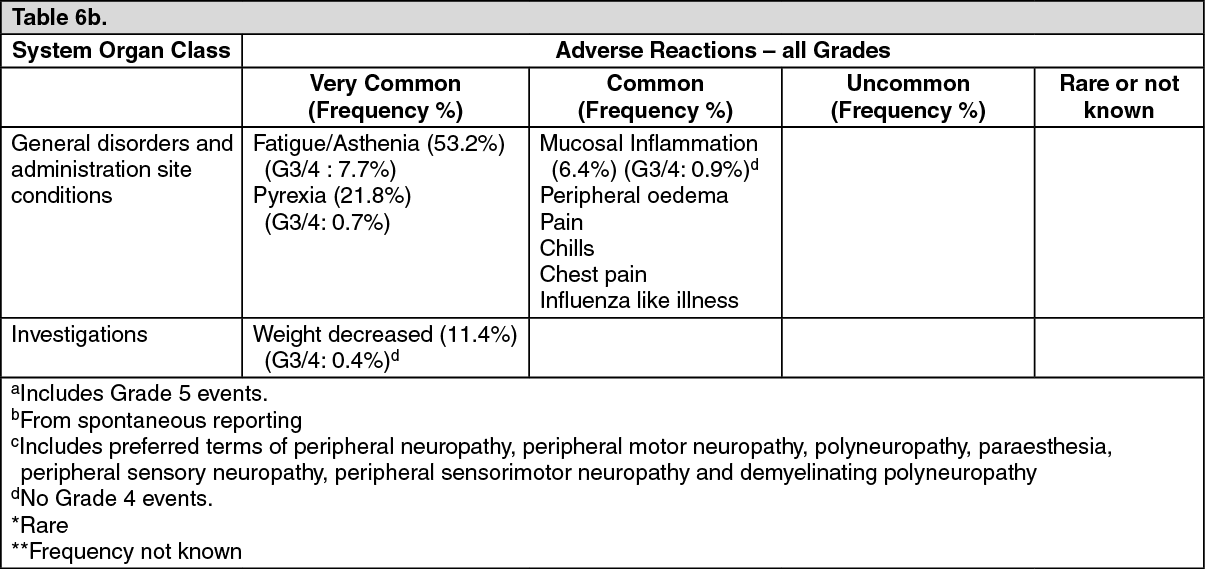 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
