Each vial contains 40 mg parecoxib (present as 42.36 mg parecoxib sodium) for reconstitution with 2 mL of solvent. After reconstitution, the final concentration of parecoxib is 20 mg/mL.
Each ampoule contains 2 mL of sodium chloride 9 mg/mL.
Excipients/Inactive Ingredients: Powder: Dibasic sodium phosphate, Phosphoric acid and/or sodium hydroxide (for pH adjustment).
Solvent: Sodium chloride, Hydrochloric Acid or sodium hydroxide (for pH adjustment), Water for Injections.
Pharmacology: Pharmacodynamics: Parecoxib is a prodrug of valdecoxib. Valdecoxib is an NSAID that exhibits anti-inflammatory, analgesic and antipyretic properties in animal models.
The mechanism of action is believed to be due to inhibition of prostaglandin synthesis primarily through inhibition of COX-2. At therapeutic plasma concentrations in humans valdecoxib does not inhibit cyclooxygenase-1 (COX-1).
Clinical Studies: Parecoxib has been studied in a broad range of major and minor surgeries. The efficacy of parecoxib was established in studies of dental, gynaecologic (hysterectomy), orthopaedic (knee and hip replacement), and coronary artery bypass graft surgical pain (see Contraindications). The first perceptible analgesic effect occurred in 7 to 13 minutes, with clinically meaningful analgesia demonstrated in 23 to 39 minutes and a peak effect within 2 hours following administration of single doses of 40 mg IV or IM parecoxib. The magnitude of analgesic effect of the 40 mg dose was comparable with that of ketorolac 60 mg IM or ketorolac 30 mg IV. After a single dose, the duration of analgesia was dose and clinical pain model dependent, and ranged from 6 to greater than 12 hours.
Use Beyond 3 Days: Most trials were designed for dosing up to 3 days. Data from 3 of 28 randomised placebo-controlled trials, where the protocols allowed treatment of parecoxib for >3 days was pooled and analysed, 358 patients received parecoxib for >3 days and 318 patients received placebo for >3 days. Both groups had similar demographics. Per the study protocols, subjects received a total of 60 mg on day one followed by 20 or 40 mg/day in the subsequent days. The mean (SD) duration of treatment was 4.1 (0.4) days for parecoxib and 4.2 (0.5) days for placebo, the range was 4 to 7 days for parecoxib and 4 to 9 days for placebo. The occurrence of AE in patients receiving parecoxib for 4 to 7 days (median duration 4 days) was low after treatment Day 3 and similar to placebo.
Opioid-sparing Effects: Parecoxib, at recommended doses, significantly reduced opioid consumption and patient-reported opioid-related adverse effects (fatigue, drowsiness, confusion, inability to concentrate, dizziness, nausea, constipation, difficult urination), while providing improved pain relief compared to opioids alone. In a placebo-controlled, orthopaedic and general surgery study (n =1050), patients received parecoxib at an initial parenteral dose of 40 mg IV followed by 20 mg twice daily for a minimum of 72 hours in addition to receiving standard care including supplemental patient controlled opioids (IV morphine sulfate). The reduction in opioid use with parecoxib treatment on Days 2 and 3 was 7.2 mg and 2.8 mg (37% and 28%, respectively). This reduction in opioid use was accompanied by significant reductions in patient-reported opioid symptom distress, as well as improved pain relief compared to opioids alone.
Additional studies in other surgical settings provided similar observations.
Platelets: In a series of small, multiple dose studies in healthy young and elderly subjects, parecoxib 20 mg or 40 mg twice daily had no effect on platelet aggregation or bleeding compared to placebo. In young subjects, Dynastat 40 mg twice daily had no clinically significant effect on acetylsalicylic acid-mediated inhibition of platelet function.
Gastrointestinal Studies: In short-term studies (7 days), the incidence of endoscopically observed gastroduodenal ulcers or erosions in healthy young and elderly (≥65 years) subjects administered parecoxib (5%-21%), although higher than placebo (5%-12%), was statistically significantly lower than the incidence observed with NSAIDs (66%-90%).
CABG Post-operative Safety Studies: In addition to routine adverse event reporting, pre-specified event categories, adjudicated by an independent expert committee, were examined in two placebo-controlled safety studies in which patients received parecoxib sodium for at least 3 days and then were transitioned to oral valdecoxib for a total duration of 10 to 14 days. All patients received standard of care analgesia during treatment.
Patients received low-dose acetylsalicylic acid prior to randomization and throughout the two CABG surgery studies.
The first CABG surgery study evaluated patients treated with IV parecoxib sodium 40 mg twice daily for a minimum of 3 days, followed by treatment with valdecoxib 40 mg twice daily (parecoxib sodium/valdecoxib group) (n=311) or placebo/placebo (n=151) in a 14-day, double-blind placebo-controlled study. Nine pre-specified adverse event categories were evaluated (cardiovascular thromboembolic events, pericarditis, new onset or exacerbation of congestive heart failure, renal failure/dysfunction, upper GI ulcer complications, major non-GI bleeds, infections, non-infectious pulmonary complications, and death). There was a significantly (p<0.05) greater incidence of cardiovascular/thromboembolic events (myocardial infarction, ischemia, cerebrovascular accident, deep vein thrombosis and pulmonary embolism) detected in the parecoxib/valdecoxib treatment group compared to the placebo/placebo treatment group for the IV dosing period (2.2% and 0.0%, respectively) and over the entire study period (4.8% and 1.3%, respectively). Surgical wound complications (most involving the sternal wound) were observed at an increased rate with parecoxib/valdecoxib treatment.
In the second CABG surgery study, four pre-specified event categories were evaluated (cardiovascular/thromboembolic; renal dysfunction/renal failure; upper GI ulcer/bleeding; surgical wound complication). Patients were randomised within 24-hours post-CABG surgery to: parecoxib initial dose of 40 mg IV, then 20 mg IV every 12 hours for a minimum of 3 days followed by valdecoxib PO (20 mg every 12 hours) (n=544) for the remainder of a 10-day treatment period; placebo IV followed by valdecoxib PO (n=544); or placebo IV followed by placebo PO (n=548). A significantly (p=0.033) greater incidence of events in the cardiovascular/thromboembolic category was detected in the parecoxib/valdecoxib treatment group (2.0%) compared to the placebo/placebo treatment group (0.5%). Placebo/valdecoxib treatment was also associated with a higher incidence of CV thromboembolic events versus placebo treatment, but this difference did not reach statistical significance. Three of the six cardiovascular thromboembolic events in the placebo/valdecoxib treatment group occurred during the placebo treatment period; these patients did not receive valdecoxib. Pre-specified events that occurred with the highest incidence in all three treatment groups involved the category of surgical wound complications, including deep surgical infections and sternal wound healing events.
There were no significant differences between active treatments and placebo for any of the other pre-specified event categories (renal dysfunction/failure, upper GI ulcer complications or surgical wound complications).
Parecoxib has not been studied in cardiovascular revascularization procedures other than CABG.
In an analysis of 17 controlled trials in non-cardiac major surgery, where the majority of patients were treated for 2 days, patients receiving parecoxib did not experience an increased risk of cardiovascular adverse events compared to placebo. This included patients with none, one or two cardiovascular risk factors. This analysis has about 77% power to detect a doubling in the background rate of cardiovascular adverse events in patients treated with parecoxib.
General Surgery: In a large (N=1050) major orthopaedic/general surgery trial, patients received an initial dose of parecoxib 40 mg IV, then 20 mg IV every 12 hours for a minimum of 3 days followed by valdecoxib PO (20 mg every 12 hours) (n=525) for the remainder of a 10-day treatment period, or placebo IV followed by placebo PO (n=525). There were no significant differences in the overall safety profile, including the four pre-specified event categories described previously for the second CABG surgery study, for parecoxib sodium/valdecoxib compared to placebo treatment in these post-surgical patients.
Pharmacokinetics: Following IV or IM injection, parecoxib is rapidly converted to valdecoxib, the pharmacologically active substance, by enzymatic hydrolysis in the liver.
Absorption: Exposure of valdecoxib following single doses of parecoxib, as measured by both the area under the plasma concentration vs. time curve (AUC) and peak concentration (Cmax), is approximately linear in the range of clinical doses. AUC and Cmax following twice daily administration is linear up to 50 mg IV and 20 mg IM. Steady-state plasma concentrations of valdecoxib were reached within 4 days with twice daily dosing.
Following single IV and IM doses of parecoxib sodium 20 mg, Cmax of valdecoxib is achieved in approximately 30 minutes and approximately 1 hour, respectively. Exposure to valdecoxib was similar in terms of AUC and Cmax following IV and IM administration. Exposure to parecoxib was similar after IV or IM administration in terms of AUC. Average Cmax of parecoxib after IM dosing was lower compared to bolus IV dosing, which is attributed to slower extravascular absorption after IM administration. These decreases were not considered clinically important since Cmax of valdecoxib is comparable after IM and IV parecoxib sodium administration.
Distribution: The volume of distribution of valdecoxib after its IV administration is approximately 55 litres.
Plasma protein binding is approximately 98% over the concentration range achieved with parecoxib 80 mg/day. Valdecoxib, but not parecoxib, is extensively partitioned into erythrocytes.
Metabolism: Parecoxib is rapidly and almost completely converted to valdecoxib and propionic acid in vivo with a plasma half-life of approximately 22 minutes. Elimination of valdecoxib is by extensive hepatic metabolism involving multiple pathways, including cytochrome P450 (CYP) 3A4 and CYP2C9 isoenzymes and glucuronidation (about 20%) of the sulphonamide moiety. A hydroxylated metabolite of valdecoxib (via the CYP pathway) has been identified in human plasma that is active as a COX-2 inhibitor. It represents approximately 10% of the concentration of valdecoxib; because of this metabolite's low concentration, it is not expected to contribute a significant clinical effect after administration of therapeutic doses of parecoxib sodium.
Elimination: Valdecoxib is eliminated via hepatic metabolism with less than 5% unchanged valdecoxib recovered in the urine. No unchanged parecoxib is detected in urine and only trace amounts in the faeces. About 70% of the dose is excreted in the urine as inactive metabolites. Plasma clearance (CLp) for valdecoxib is about 6 l/hr. After IV or IM dosing of parecoxib sodium, the elimination half-life (t1/2) of valdecoxib is about 8 hours.
Elderly Subjects: Dynastat has been administered to 335 elderly patients (65-96 years of age) in pharmacokinetic and therapeutic trials. In healthy elderly subjects, the apparent oral clearance of valdecoxib was reduced, resulting in an approximately 40% higher plasma exposure of valdecoxib compared to healthy young subjects. When adjusted for body weight, steady state plasma exposure of valdecoxib was 16% higher in elderly females compared to elderly males.
Renal Impairment: In patients with varying degrees of renal impairment administered 20 mg IV Dynastat, parecoxib was rapidly cleared from plasma. Because renal elimination of valdecoxib is not important to its disposition, no changes in valdecoxib clearance were found even in patients with severe renal impairment or in patients undergoing dialysis.
Hepatic Impairment: Moderate hepatic impairment did not result in a reduced rate or extent of parecoxib conversion to valdecoxib. In patients with moderate hepatic impairment (Child-Pugh Class B), treatment should be initiated at the lowest recommended dose of Dynastat and the maximum daily dose should be reduced to 40 mg since valdecoxib exposures were more than doubled (130%) in these patients.
Patients with severe hepatic impairment have not been studied and therefore the use of Dynastat in patients with severe hepatic impairment is contraindicated (see Dosage & Administration and Contraindications).
Toxicology: Preclinical safety data: There were no findings of teratogenicity in studies in rats and rabbits. Studies in rats at maternally toxic doses and studies in rabbits at the maximal evaluable dose have not revealed embryotoxic effects other than post-implantation loss, which has been observed with other drugs that inhibit prostaglandin synthesis.
Parecoxib, valdecoxib (its active metabolite) and a valdecoxib active metabolite are excreted in the milk of lactating rats.
Parecoxib is indicated for the short-term treatment of acute post-operative pain.
There is limited clinical experience with parecoxib treatment beyond three days.
Parecoxib may be administered as single or multiple IV or IM doses on a regular or as needed schedule. After initiation of therapy, dosage should be adjusted based on patient response.
Parecoxib is only indicated for patients with a need for parenteral therapy and for whom a similar benefit could not be obtained from alternative oral therapy. It is recommended that patients be transitioned to alternative oral therapy as soon as clinically indicated.
As the cardiovascular (CV) risk of cyclooxygenase-2 (COX-2) specific inhibitors may increase with dose and duration of exposure, the shortest duration possible and the lowest effective daily dose should be used.
Management of Acute Pain: The recommended single or initial dose for treatment of acute pain is 40 mg, administered either IV or IM, followed by 20 mg every 6 to 12 hours, as required. The maximum daily dosage is 60 mg. The IV bolus injection may be given directly into a vein or into an existing IV line. The IM injection should be given slowly and deeply into the muscle.
Concomitant Use with Opioid Analgesics: Opioid analgesics can be used concurrently with parecoxib, dosing as described previously. In clinical trials, the daily requirement for opioids was significantly reduced (20%-40%) when co-administered with parecoxib. An optimal effect is achieved when parecoxib is given prior to opioid administration. In all clinical assessments parecoxib was administered at a fixed time interval whereas the opioids were administered on as needed basis (PRN).
Elderly: No dosage adjustment is generally necessary. However, for elderly patients weighing less than 50 kg, it is advisable to reduce the initial dose of parecoxib by 50%. The maximum daily dose should be reduced to 40 mg in elderly patients weighing less than 50 kg.
Hepatic Impairment: No dosage adjustment is necessary in patients with mild hepatic impairment (Child-Pugh Class A). Treatment with parecoxib should be initiated at the lowest recommended dose and the maximum daily dose should be reduced to 40 mg in patients with moderate hepatic impairment (Child-Pugh Class B).
Patients with severe hepatic impairment (Child-Pugh Class C) have not been studied. The use of parecoxib in these patients is contraindicated (see Contraindications and Pharmacology: Pharmacokinetics under Actions).
Renal Impairment: In patients with severe renal impairment (creatinine clearance <30 mL/minute), or patients who may be predisposed to fluid retention, parecoxib should be initiated at the lowest recommended dose and the patient's kidney function closely monitored.
Co-administration with Fluconazole: When parecoxib is co-administered with fluconazole, the lowest recommended dose of parecoxib should be used.
Paediatric Patients: Safety and efficacy have not been established in children under 18 years of age. Therefore, parecoxib is not recommended in these patients.
Clinical experience of overdose is limited. Single IV doses of up to 200 mg parecoxib have been administered to healthy subjects without clinically significant adverse effects. Parecoxib doses of 50 mg IV twice daily (100 mg/day) for 7 days did not result in any signs of toxicity.
In case of suspected acute overdose, appropriate supportive and symptomatic medical care should be provided. There are no specific antidotes. Dialysis is unlikely to be an efficient method of drug removal, because of high protein binding of the drug.
Parecoxib is contraindicated in: patients with known hypersensitivity to parecoxib or to any other ingredient of the product.
History of previous serious allergic drug reaction of any type, especially cutaneous reaction such as Stevens-Johnson syndrome, toxic epidermal necrolysis, erythema multiforme or patients with known hypersensitivity to sulphonamides (see Precautions and Adverse Reactions).
Active peptic ulceration or gastrointestinal (GI) bleeding.
Patients who have experienced bronchospasm, acute rhinitis, nasal polyps, angioneurotic oedema, urticaria, or other allergic-type reactions after taking acetylsalicylic acid (aspirin) or non-steroidal anti-inflammatory drugs (NSAIDs), including other COX-2 specific inhibitors.
The third trimester of pregnancy and breast-feeding.
Severe hepatic impairment (serum albumin <25 g/L or Child-Pugh Class C).
Inflammatory bowel disease.
Congestive heart failure (NYHA II-IV).
Treatment of post-operative pain following coronary artery bypass graft (CABG) surgery.
Established ischemic heart disease, peripheral arterial disease and/or cerebrovascular disease.
Administration other than IV or IM: Modes of administration other than IV or IM (e.g., intra-articular, intrathecal) have not been studied and should not be used.
Because of the possibility for increased adverse reactions at higher doses of parecoxib, other COX-2 inhibitors and NSAIDs, patients treated with parecoxib should be reviewed following dose increase and, in the absence of an increase in efficacy, other therapeutic options should be considered. There is limited clinical experience with Dynastat treatment beyond three days.
If, during treatment, patients deteriorate in any of the organ system functions described as follows, appropriate measures should be taken and discontinuation of parecoxib therapy should be considered.
Cardiovascular Effects: COX-2 inhibitors, of which parecoxib is one, have been associated with an increased risk of cardiovascular and thrombotic adverse events when taken long term. The relative increase of this risk appears to be similar in those with or without known CV disease or CV risk factors. However, patients with known cardiovascular disease or CV risk factors may be at greater risk in terms of absolute incidence, due to their increased rate at baseline. The exact magnitude of the risk associated with a single dose has not been determined, nor has the exact duration of therapy associated with increased risk.
Patients with significant risk factors for cardiovascular events (e.g., hypertension, hyperlipidaemia, diabetes mellitus, smoking) should only be treated with parecoxib after careful consideration.
Appropriate measures should be taken and discontinuation of parecoxib therapy should be considered if there is clinical evidence of deterioration in the condition of specific clinical symptoms in these patients. Dynastat has not been studied in cardiovascular revascularization procedures other than CABG procedures.
Two separate studies in coronary artery bypass graft (CABG) surgery showed that patients receiving parecoxib for a minimum of 3 days followed by oral valdecoxib (the active metabolite of parecoxib) for 7 to 14 days, had increased incidence of cardiovascular/thromboembolic events (e.g., myocardial infarction and cerebrovascular accident) compared to those receiving placebo (see Pharmacology: Pharmacodynamics under Actions). Parecoxib is therefore contraindicated for the treatment of post-operative pain immediately following CABG surgery.
Gastrointestinal (GI) Effects: Upper gastrointestinal (GI) perforations, ulcers, or bleeds, some of them resulting in fatal outcome, have occurred in patients treated with parecoxib. Patients most at risk of developing these types of GI complications with NSAIDs are the elderly, patients with cardiovascular disease, or patients with a history of, or active, GI disease, such as ulceration, bleeding, or inflammatory conditions; or patients using concomitant aspirin. The NSAIDs class is also associated with increased GI complications when co-administered with corticosteroids, selective serotonin reuptake inhibitors, other antiplatelet drugs, other NSAIDs or patients ingesting alcohol, however, there are currently no specific parecoxib clinical data.
Skin Effects: Valdecoxib, the active moiety of parecoxib, contains a sulphonamide moiety and patients with a known history of a sulphonamide allergy may be at a greater risk of skin reactions. Patients without a history of sulphonamide allergy may also be at risk for serious skin reactions.
Serious skin reactions, including erythema multiforme, exfoliative dermatitis and Stevens-Johnson syndrome (some of them fatal) have been reported through post-marketing surveillance in patients receiving parecoxib. In addition to erythema multiforme and Stevens-Johnson syndrome, fatal reports of toxic epidermal necrolysis have been reported through post-marketing surveillance in patients receiving valdecoxib and the potential cannot be ruled out for parecoxib. Generalised bullous fixed drug eruption (GBFDE) may occur with parecoxib exposure based on a reaction with etoricoxib exposure.
Drug reaction with eosinophilia and systemic symptoms syndrome (DRESS syndrome) may occur with parecoxib exposure based on other serious skin reactions reported with celecoxib and valdecoxib exposure.
Patients appear to be at highest risk for these events early in the course of therapy, with the onset of the event occurring in the majority of cases within the first two weeks of treatment.
Appropriate measures should be taken by physicians to monitor for any serious skin reactions with therapy, e.g., additional patient consultations. Patients should be advised to immediately report any emergent skin condition to their physician.
Parecoxib should be discontinued at the first appearance of skin rash, mucosal lesions or any other sign of hypersensitivity. Serious skin reactions have been reported with other COX-2 inhibitors during post-marketing experience. The reported rate of these events appears to be greater for valdecoxib as compared to other COX-2 agents.
Anaphylactoid Reactions: Hypersensitivity reactions (anaphylactic reactions and angioedema) have been reported in post-marketing experience with valdecoxib and parecoxib (see Adverse Reactions). These reactions have occurred in patients with and without a history of allergic-type reactions to sulphonamides (see Contraindications). Parecoxib should be discontinued at the first sign of hypersensitivity.
Severe Hypotension: Cases of severe hypotension shortly following parecoxib administration have been reported in post-marketing experience with parecoxib. Some of these cases have occurred without other signs of anaphylaxis. The practitioner should be prepared to treat severe hypotension.
Use with Oral Anticoagulants: The concomitant use of NSAIDs with oral anticoagulants increases the risk of bleeding. Oral anticoagulants include warfarin/coumarin-type and novel oral anticoagulants (e.g., apixaban, dabigatran, and rivaroxaban).
Co-administration of parecoxib with warfarin caused a small increase in the AUC of warfarin, and also in the prothrombin time (measured by International Normalised Ratio [INR]). While mean INR values were only slightly increased with co-administration of parecoxib, the day-to-day variability in individual INR values was increased. Anticoagulant activity should be monitored, particularly during the first few days after initiating parecoxib, in patients receiving warfarin or similar agents, since these patients may be at increased risk of bleeding complications.
Hypertension: As with all NSAIDs, parecoxib can lead to the onset of new hypertension or worsening of pre-existing hypertension, either of which may contribute to the increased incidence of cardiovascular events. NSAIDs, including parecoxib, should be used with caution in patients with hypertension. Blood pressure should be monitored closely during the initiation of therapy with parecoxib and throughout the course of therapy. If blood pressure rises significantly, alternative treatment should be considered.
Fluid Retention and Oedema: As with other drugs known to inhibit prostaglandin synthesis, fluid retention and oedema have been observed in some patients taking parecoxib. Therefore, parecoxib should be used with caution in patients with compromised cardiac function, pre-existing oedema, or other conditions pre-disposing to, or worsened by, fluid retention including those taking diuretic treatment or otherwise at risk of hypovolemia. If there is clinical evidence of deterioration in the condition of these patients, appropriate measures including discontinuation of parecoxib should be taken.
Renal Effects: Acute renal failure has been reported through post-marketing surveillance in patients receiving parecoxib (see Adverse Reactions). Since prostaglandin synthesis inhibition may result in deterioration of renal function and fluid retention, caution should be observed when administering Dynastat in patients with impaired renal function or hypertension, or in patients with compromised cardiac or hepatic function or other conditions pre-disposing to fluid retention. Renal function should be closely monitored in patients with advanced renal disease who are administered parecoxib (see Dosage & Administration).
Caution should be used when initiating treatment in patients with dehydration. It is advisable to rehydrate patients first and then start therapy with parecoxib.
Hepatic Effects: Patients with severe hepatic impairment (Child-Pugh Class C) have not been studied. The use of parecoxib in patients with severe hepatic impairment is contraindicated. Parecoxib should be used with caution when treating patients with moderate hepatic impairment (Child-Pugh Class B), and initiated at the lowest recommended dose (see Dosage & Administration).
A patient with symptoms and/or signs of liver dysfunction, or in whom an abnormal liver function test has occurred, should be monitored carefully for evidence of the development of a more severe hepatic reaction while on therapy with parecoxib.
General: By reducing inflammation, parecoxib may diminish the utility of diagnostic signs, such as fever, in detecting infections. The concomitant use of parecoxib with other non-specific NSAIDs should be avoided.
Effects on ability to drive and use machines: Patients who experience dizziness, vertigo or somnolence after receiving Dynastat should refrain from driving or operating machines.
Fertility: Based on the mechanism of action, the use of NSAIDs may delay or prevent rupture of ovarian follicles, which has been associated with reversible infertility in some women. In women who have difficulties conceiving or who are undergoing investigation of infertility, withdrawal of NSAIDs, including parecoxib, should be considered.
Pregnancy: Parecoxib is suspected to cause serious birth defects when administered during the last trimester of pregnancy because as with other medicinal products known to inhibit prostaglandin, it may cause premature closure of the ductus arteriosus or uterine inertia (see Contraindications and Pharmacology: Pharmacodynamics under Actions).
Dynastat is contraindicated (see Contraindications) in the last trimester of pregnancy.
Inhibition of prostaglandin synthesis might adversely affect pregnancy. Data from epidemiological studies suggest an increased risk of spontaneous abortion after use of prostaglandin synthesis inhibitors in early pregnancy. In animals, administration of prostaglandin synthesis inhibitors has been shown to result in increased pre- and post-implantation loss (see Pharmacology: Toxicology: Preclinical safety data under Actions).
Dynastat is not recommended in women attempting to conceive (see Precautions and Pharmacology: Pharmacodynamics under Actions).
There are no adequate data from the use of parecoxib in pregnant women or during labour. Studies in animals have shown reproductive toxicity (see Pharmacology: Pharmacodynamics under Actions). The potential risk for humans is unknown. Dynastat should not be used during the first two trimesters of pregnancy unless clearly necessary (i.e., the potential benefit to the patient outweighs the potential risk to the foetus).
Use of NSAIDs at about 20 weeks gestation or later in pregnancy may cause foetal renal dysfunction leading to oligohydramnios and in some cases, neonatal renal impairment. These adverse outcomes are seen, on average, after days to weeks of treatment, although oligohydramnios has been infrequently reported as soon as 48 hours after NSAID initiation. Oligohydramnios is often, but not always, reversible with treatment discontinuation.
Lactation: Administration of a single dose of parecoxib to lactating women resulted in the transfer of a relatively small amount of parecoxib and its active metabolite into breast milk, and this resulted in a low relative dose for the infant (less than 1% of the weight-adjusted maternal dose). Dynastat must not be administered to women who breast-feed (see Contraindications).
The most common adverse reaction for Dynastat is nausea. The most serious reactions occur uncommonly to rarely, and include cardiovascular events such as myocardial infarction and severe hypotension, as well as hypersensitivity events such as anaphylaxis, angioedema and severe skin reactions. Following coronary artery bypass graft surgery, patients administered Dynastat have a higher risk of adverse reactions such as: cardiovascular/thromboembolic events (including myocardial infarction, stroke/TIA, pulmonary embolus, and deep vein thrombosis; see Contraindications and Pharmacology: Pharmacodynamics under Actions), deep surgical infections, and sternal wound healing complications.
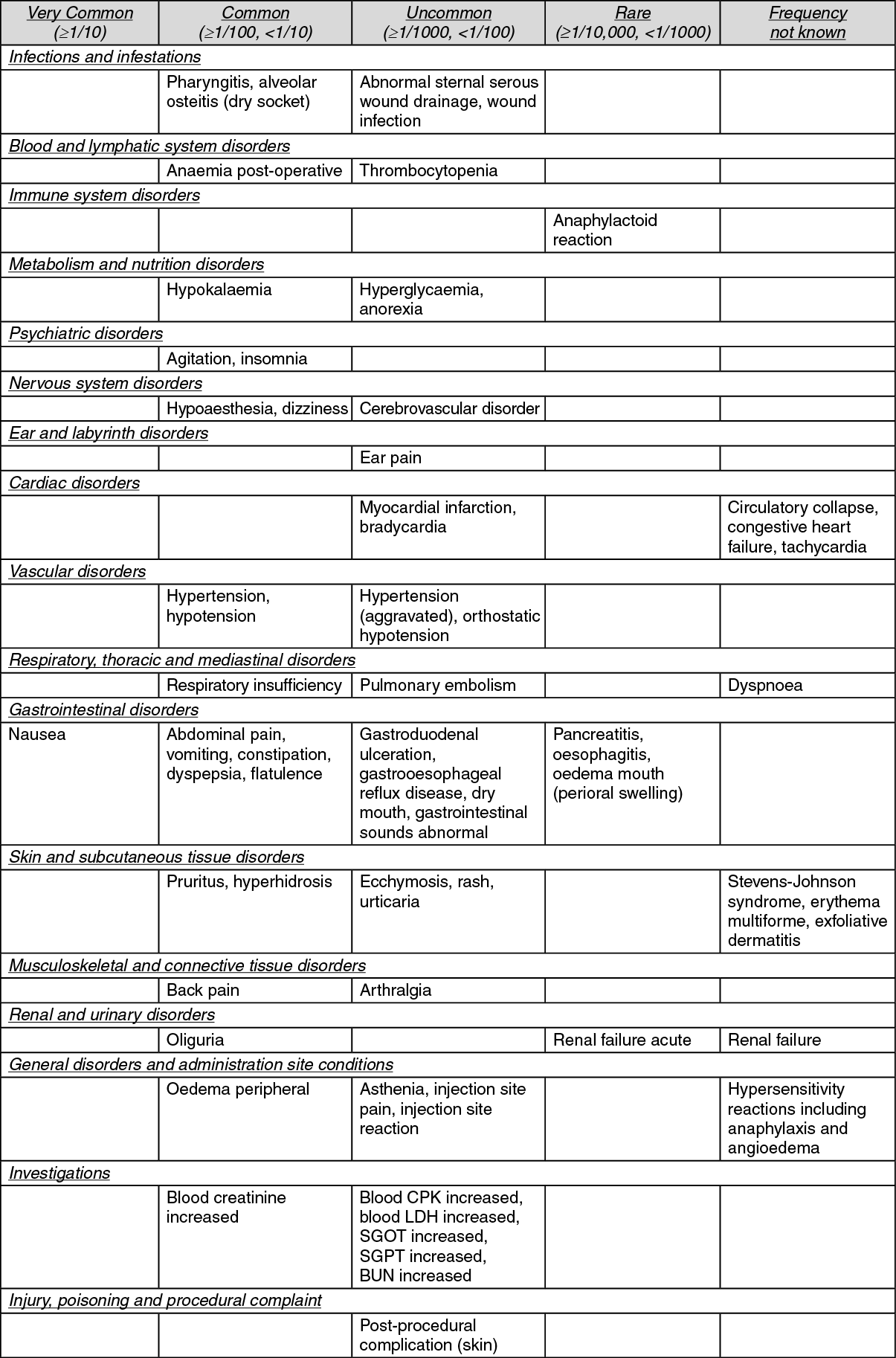
Tabulated list of adverse reactions: The following adverse reactions were reported for patients who received parecoxib (N=5,402) in 28 placebo-controlled clinical trials. Reports from post-marketing experience have been listed as "frequency not known" because the respective frequencies cannot be estimated from the available data. Within each frequency grouping, adverse reactions are listed using MedDRA terminology and presented in order of decreasing seriousness. (See table.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In post-marketing experience, in addition to the severe cutaneous adverse reaction erythema multiforme and Stevens-Johnson's syndrome, toxic epidermal necrolysis has been reported in association with the use of valdecoxib, and cannot be ruled out for parecoxib (see Precautions). In addition, the following rare, serious adverse reactions have been reported in association with the use of NSAIDs and cannot be ruled out for Dynastat: bronchospasm and hepatitis.
General: The drug interaction studies were performed with either parecoxib or the active moiety (valdecoxib).
In humans, parecoxib undergoes extensive hepatic metabolism involving P450 isoenzymes 3A4 and 2C9, and non-P450 dependent pathways (i.e., glucuronidation). Concomitant administration of parecoxib with known CYP 3A4 and 2C9 inhibitors can result in increased AUC of parecoxib.
Drug-Specific: Interaction of parecoxib with warfarin or similar agents: See Precautions.
Fluconazole and ketoconazole: Co-administration of fluconazole, a CYP2C9 inhibitor, and ketoconazole, a CYP3A4 inhibitor, enhanced the AUC of valdecoxib by 62% and 38%, respectively. When parecoxib is co-administered with fluconazole, the lowest recommended dose of parecoxib should be used. No dosage adjustment is necessary when parecoxib is co-administered with ketoconazole (see Dosage & Administration).
Anti-hypertensives including ACE-inhibitors, angiotensin II antagonists, beta blockers and diuretics: Inhibition of prostaglandins may diminish the effect of angiotensin converting enzyme (ACE) inhibitors, angiotensin II antagonists, beta-blockers and diuretics. This interaction should be given consideration in patients receiving parecoxib concomitantly with ACE-inhibitors, angiotensin II antagonists, beta-blockers and diuretics.
In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, co-administration of NSAIDs, including selective COX-2 inhibitors, with ACE inhibitors and/or angiotensin II antagonists, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible.
Therefore, the concomitant administration of these drugs should be done with caution. Patients should be adequately hydrated and the need to monitor the renal function should be assessed at the beginning of the concomitant treatment and periodically thereafter.
Diuretics: Clinical studies have shown that NSAIDs, in some patients, can reduce the natriuretic effect of furosemide and thiazides by inhibition of renal prostaglandin synthesis.
Cyclosporine: Because of their effect on renal prostaglandins, NSAIDs may increase the risk of nephrotoxicity with cyclosporine.
Methotrexate: A pharmacokinetic interaction study was conducted using valdecoxib and methotrexate and no clinically important interactions were seen. However, caution is advised when methotrexate is administered concurrently with NSAIDs, because NSAID administration may result in increased plasma levels of methotrexate.
Lithium: Valdecoxib produced significant decreases in lithium serum clearance (25%) and renal clearance (30%) resulting in a 34% higher serum AUC compared to lithium alone. Lithium serum concentrations should be monitored closely when initiating or changing parecoxib therapy in patients receiving lithium.
Other: Interaction studies were conducted between parecoxib and IV or oral midazolam, heparin, propofol, fentanyl, and alfentanil. Interaction studies were also conducted between valdecoxib and glibenclamide (glyburide), oral contraceptives (ethinyl oestradiol/norethindrone), phenytoin, omeprazole and diazepam. No clinically important interactions were seen in these studies.
Parecoxib may be co-administered with opioid analgesics. In clinical trials, the daily requirement for PRN opioids was significantly reduced when co-administered with parecoxib.
No formal interaction studies were performed with parecoxib and inhalation anaesthetic agents, such as nitrous oxide and isoflurane; however, no evidence of a drug interaction was observed in clinical studies.
Parecoxib does not interfere with the anti-platelet effect of low-dose aspirin. Clinical trials indicate that parecoxib can be given with low-dose aspirin (≤325 mg). In the submitted studies, as with other NSAIDs, an increased risk of gastrointestinal ulceration or other gastrointestinal complications compared to use of parecoxib alone was shown for concomitant administration of low-dose aspirin. Because of its lack of platelet effects, parecoxib is not a replacement for aspirin in the prophylactic treatment of cardiovascular disease.
Co-administration of NSAIDs and cyclosporine or tacrolimus has been suggested to increase the nephrotoxic effect of cyclosporine and tacrolimus. Renal function should be monitored when parecoxib and any of these medicinal products are co-administered.
Treatment with valdecoxib (40 mg twice daily for 7 days) produced a 3-fold increase in plasma concentrations of dextromethorphan (CYP2D6 substrate). Therefore, caution should be observed when co-administering parecoxib and medicinal products that are predominantly metabolised by CYP2D6 and which have narrow therapeutic margins (e.g., flecainide, propafenone, metoprolol).
Incompatibilities: Following reconstitution with an acceptable diluent, parecoxib sodium may be injected into an IV line delivering 0.9% Sodium Chloride Injection, 5% Dextrose Injection, Lactated Ringers Injection, or 5% Dextrose and 0.45% Sodium Chloride Injection. Injection into a line delivering 5% Dextrose in Lactated Ringer's, or other IV fluid not listed here, is not recommended, as this may cause precipitation from solution.
Parecoxib sodium should not be admixed for injection with any other drug.
Do not inject parecoxib into an IV line delivering any other drug. The IV line must be adequately flushed prior to, and after parecoxib injection with a solution of known compatibility (see Special precautions for disposal and other handling as follows).
Special precautions for disposal and other handling: Parecoxib sodium for injection is a preservative-free lyophilised powder. Parecoxib sodium should be reconstituted with 1 mL (20 mg vial) or 2 mL (40 mg vial) Sodium Chloride Injection (0.9%).
Alternatively, parecoxib sodium may be reconstituted with bacteriostatic 0.9% Sodium Chloride Injection, 5% Dextrose Injection or 5% Dextrose and 0.45% Sodium Chloride Injection.
Use of Lactated Ringer's Injection, or 5% Dextrose in Lactated Ringer's Injection, are not recommended for reconstitution as they will cause the drug to precipitate from solution. Use of Water for Injection is not recommended for reconstitution of parecoxib sodium, as the resulting solution is not isotonic.
Do not refrigerate or freeze the reconstituted product.
Shelf life: Reconstituted Solution: Chemical and physical in-use stability has been demonstrated for 48 hours at 30°C.
From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at the 2°C to 8°C, unless reconstitution has been taken place in controlled and validated aseptic conditions.
Store at or below 30°C.
Do not refrigerate or freeze reconstituted solution.
For single use only. Any unused solution, solvent or waste material should be disposed of according to local requirements.
M01AH04 - parecoxib ; Belongs to the class of non-steroidal antiinflammatory and antirheumatic products, coxibs.
Dynastat powd for inj 40 mg
(vial + 2-mL amp solvent of NaCl 9 mg/mL soln) 5 × 1's



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
