


 Sign Out
Sign Out




Pharmacotherapeutic group: Cardiac therapy. ATC code: C01DX22.
Pharmacology: Mechanism of Action: Vericiguat is a stimulator of soluble guanylate cyclase (sGC). Heart failure is associated with impaired synthesis of nitric oxide (NO) and decreased activity of its receptor, sGC. Soluble guanylate cyclase catalyzes synthesis of intracellular cyclic guanosine monophosphate (cGMP), an important signaling molecule that regulates critical physiological processes such as cardiac contractility, vascular tone, and cardiac remodeling. Deficiency in sGC-derived cGMP contributes to myocardial and vascular dysfunction. Vericiguat restores the relative deficiency in this signaling pathway by directly stimulating sGC, independently of and synergistically with NO, to augment the levels of intracellular cGMP, which may improve both myocardial and vascular function. The complementary cardiovascular benefits of vericiguat in heart failure patients are therefore attributed to the active restoration of the deficient NO-sGCcGMP pathway driving heart failure progression.
Pharmacodynamics: The pharmacodynamic effects of vericiguat were evaluated after single and multiple dose administrations in healthy subjects and in patients with heart failure and are consistent with the mode of action of an sGC stimulator resulting in smooth muscle relaxation and vasodilation. Over the course of the VICTORIA study, the mean reduction in systolic blood pressure was approximately 1 to 2 mmHg greater in patients who received Vericiguat (Verquvo) compared with placebo.
In a 12-week placebo-controlled dose-finding study (SOCRATES-REDUCED) in patients with heart failure, vericiguat demonstrated a dose-dependent reduction in NT-proBNP, a biomarker in heart failure, compared to placebo when added to standard of care. In VICTORIA, the estimated reduction from baseline NT-proBNP at week 32 was greater in patients who received Vericiguat (Verquvo) compared with placebo [see Clinical Studies as follows].
Cardiac Electrophysiology: There was no evidence of proarrhythmic risk in an in vitro assessment of vericiguat or its major N glucuronide metabolite. No inhibition of cardiac ion channels (hERG, hNav1.5, or hKvLQT1/mink) was observed at substantial multiples of their unbound Cmax values at the recommended target dose of 10 mg.
The integrated risk assessment of nonclinical and clinical data supports that administration of vericiguat 10 mg is not associated with clinically meaningful QTc prolongation.
Clinical Studies: VICTORIA was a randomized, parallel-group, placebo-controlled, double-blind, event-driven, multi-center trial comparing Vericiguat (Verquvo) and placebo in 5,050 adult patients with symptomatic chronic heart failure (New York Heart Association [NYHA] class II-IV) and left ventricular ejection fraction (LVEF) less than 45% following a worsening heart failure event.
A worsening heart failure event was defined as heart failure hospitalization within 6 months before randomization or use of outpatient IV diuretics for heart failure within 3 months before randomization.
The primary objective of VICTORIA was to determine whether Vericiguat (Verquvo) in combination with other heart failure therapies is superior to placebo in reducing the risk of cardiovascular (CV) death or heart failure hospitalization in adults with symptomatic chronic heart failure and ejection fraction less than 45% following a worsening heart failure event.
Patients were treated up to the target maintenance dose of Vericiguat (Verquvo) 10 mg once daily or matching placebo. Therapy was initiated at Vericiguat (Verquvo) 2.5 mg once daily and increased in approximately 2-week intervals to 5 mg once daily and then 10 mg once daily, as tolerated. After approximately 1 year, 90% of patients in both the Vericiguat (Verquvo) and placebo arms were treated with the 10 mg target dose.
The primary endpoint was the time to first event of the composite of CV death or hospitalization for heart failure. The median follow-up for the primary endpoint was 11 months.
The population was 64% Caucasian, 22% Asian, and 5% Black. The mean age was 67 years and 76% were male. At randomization, 59% of patients were NYHA Class II, 40% were NYHA Class III, and 1% were NYHA Class IV. The mean left ventricular ejection fraction (EF) was 29% and approximately half of all patients had an EF <30%, and 14% of patients had an EF between 40% and 45%. The most frequently reported medical history conditions other than heart failure included hypertension (79%), coronary artery disease (58%), hyperlipidemia (57%), diabetes mellitus (47%), atrial fibrillation (45%), and myocardial infarction (42%). At randomization, the mean eGFR was 62 mL/min/1.73 m2; the majority of patients (88%) had an eGFR >30 mL/min/1.73 m2, and 10% of patients had an eGFR ≤30 mL/min/1.73 m2.
Sixty-seven percent of the patients in VICTORIA were enrolled within 3 months of a HF hospitalization index event; 17% were enrolled within 3 to 6 months of HF hospitalization, and 16% were enrolled within 3 months of outpatient treatment with IV diuretics for worsening HF.
The median NT-proBNP level was 2816 pg/mL at randomization.
At baseline, more than 99% of patients were treated with other heart failure therapies; 93% of patients were on a beta blocker, 73% of patients were on an angiotensin-converting enzyme (ACE) inhibitor or angiotensin II receptor blocker (ARB), 70% of patients were on a mineralocorticoid receptor antagonist (MRA), 15% of patients were on a combination of an angiotensin receptor and neprilysin inhibitor (ARNI), 28% of patients had an implantable cardiac defibrillator, and 15% had a biventricular pacemaker. Ninety-one percent of patients were treated with 2 or more heart failure medications (beta blocker, any renin-angiotensin system [RAS] inhibitor, or MRA) and 60% of patients were treated with all 3. At baseline, 6% of patients were on ivabradine and 3% of patients were on a sodium glucose co-transporter 2 (SGLT2) inhibitor.
In VICTORIA, Vericiguat (Verquvo) was superior to placebo in reducing the risk of CV death or heart failure hospitalization based on a time-to-event analysis (hazard ratio [HR]: 0.90, 95% confidence interval [CI], 0.82-0.98; p=0.019). Over the course of the study, there was a 4.2% annualized absolute risk reduction (ARR) with Vericiguat (Verquvo) compared with placebo.
Therefore, 24 patients would need to be treated over an average of 1 year to prevent 1 primary endpoint event. The treatment effect reflected a reduction in both cardiovascular death and heart failure hospitalization; see Table 1.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe Kaplan-Meier curve (Figure 1) shows time to first occurrence of the primary composite endpoint of cardiovascular death or heart failure hospitalization. (See Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn VICTORIA, Vericiguat (Verquvo) was superior to placebo in reducing the risk of all-cause mortality or HF hospitalization (HR 0.90 [95% CI, 0.83-0.98]) and total events (first and recurrent) of HF hospitalization (HR 0.91 [95% CI, 0.84-0.99]); see Tables 2 and 3. The total number of HF hospitalization events was greater in the placebo group (1,336 events) than the Vericiguat (Verquvo) group (1,223 events). (See Tables 2 and 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA wide range of demographic characteristics, baseline disease characteristics, and baseline concomitant medications were examined for their influence on outcomes. The results of the prespecified subgroup analysis for the primary composite endpoint are shown in Figure 2. (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: General Introduction: Vericiguat shows slightly less than dose proportional, time-independent pharmacokinetics, with low to moderate variability when administered with food. Vericiguat accumulates in plasma up to 155-171% and reaches pharmacokinetic steady-state after approximately 6 days. The mean steady-state population pharmacokinetic (PK) parameters of vericiguat in heart failure patients are summarized in Table 4. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAbsorption: The absolute bioavailability of vericiguat is high (93%) when taken with food. Bioavailability (AUC) and peak plasma levels (Cmax) of vericiguat administered orally as a crushed tablet in water is comparable to that of a whole tablet [see Adults under Dosage & Administration].
Effects of Food: Administration of vericiguat with a high-fat, high-calorie meal increases Tmax from about 1 hour (fasted) to about 4 hours (fed), reduces PK variability, and increases vericiguat exposure by 19% (AUC) and 9% (Cmax) for the 5 mg tablet and by 44% (AUC) and 41% (Cmax) for the 10 mg tablet as compared with the fasted state. Similar results were obtained when vericiguat was administered with a low-fat, high-carbohydrate meal. Therefore, Vericiguat (Verquvo) should be taken with food [see Adults under Dosage & Administration].
Distribution: The mean steady-state volume of distribution of vericiguat in healthy subjects is approximately 44 L. Plasma protein binding of vericiguat is about 98%, with serum albumin being the main binding component. Plasma protein binding of vericiguat is not altered by renal or hepatic impairment.
Metabolism: Glucuronidation is the major biotransformation pathway of vericiguat to form an N-glucuronide, which is pharmacologically inactive and the major drug related component in plasma. N-glucuronidation is catalyzed predominantly by UGT1A9, as well as UGT1A1. CYP mediated metabolism is a minor clearance pathway (<5%).
Elimination: Vericiguat is a low-clearance drug (1.6 L/h in healthy subjects). The half-life is about 20 hours in healthy subjects and 30 hours in heart failure patients. Following oral administration of [14C]-vericiguat to healthy subjects, approximately 53% of the dose was excreted in urine (primarily as the N-glucuronide) and 45% of the dose was excreted in feces (primarily as vericiguat).
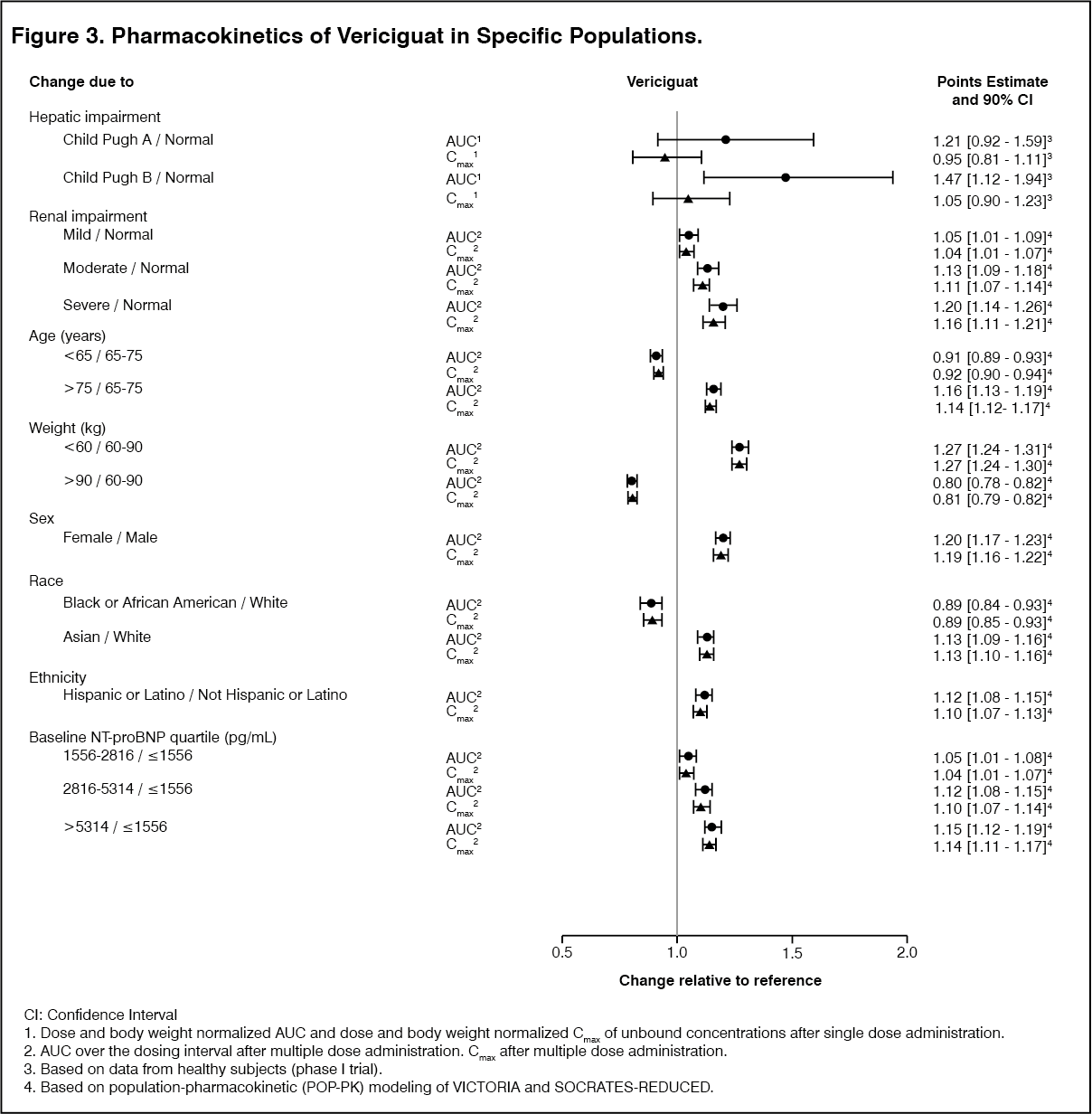
Special Populations: Effects of specific populations on the pharmacokinetics of vericiguat are shown in Figure 3. (See Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageRenal Impairment: No relevant increase in exposure (AUC) was observed for heart failure patients with moderate and severe renal impairment not requiring dialysis. In patients with heart failure with moderate (eGFR ≥30 to <60 mL/min/1.73m2) and severe renal impairment (eGFR ≥15 to <30 mL/min/1.73m2) not requiring dialysis, the mean exposure (AUC) of vericiguat was increased by 13% and 20%, respectively, compared to patients with normal renal function.
The pharmacokinetics of vericiguat have not been studied in patients with eGFR <15 mL/min/1.73m2 at treatment initiation or on dialysis [see Renal Impairment under Dosage & Administration and Renal Impairment under Precautions].
Hepatic Impairment: No relevant increase in exposure (unbound AUC) was observed for subjects with mild hepatic impairment (Child Pugh A) with mean exposure to vericiguat 21% higher compared to healthy subjects with normal hepatic function. In subjects with moderate hepatic impairment (Child Pugh B), mean exposure to vericiguat was approximately 47% higher compared to their healthy subjects with normal hepatic function. The pharmacokinetics of vericiguat have not been studied in patients with severe hepatic impairment (Child-Pugh C) [see Hepatic Impairment under Dosage & Administration and Hepatic Impairment under Precautions].
Pediatric: No studies with Vericiguat (Verquvo) have been performed in pediatric patients.
Body Weight: In a population pharmacokinetic analysis of vericiguat, the steady-state AUC values were approximately 27% higher in heart failure patients with a body weight <60 kg and approximately 20% lower in heart failure patients with a body weight >90 kg, compared to heart failure patients with a body weight between 60 and 90 kg. The effect of body weight on vericiguat exposure is not clinically meaningful.
Effects of Age, Gender, Ethnicity, Race, and Baseline NT-proBNP: Based on a population pharmacokinetic analysis, age, gender, ethnicity, race, and baseline NT-proBNP do not have a clinically meaningful effect on the pharmacokinetics of vericiguat.
Drug Interaction Studies: In Vitro Assessment of Drug Interactions: In vitro studies indicate that vericiguat and its N-glucuronide are neither inhibitors of major CYP isoforms (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4) or UGT isoforms (UGT1A1, 1A4, 1A6, 1A9, 2B4, and 2B7), nor inducers of CYP1A2, 2B6, and 3A4, at clinically relevant concentrations.
Vericiguat is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) transporters and is not a substrate of organic cation transporter (OCT1), or organic anion transporting polypeptides (OATP1B1 and OATP1B3). Vericiguat and its N-glucuronide are not inhibitors of drug transporters, including P-gp, BCRP, BSEP, OATP1B1/1B3, OAT1, OAT3, OCT1, OCT2, MATE1, and MATE2K, at clinically relevant concentrations.
Overall, these data indicate that the administration of Vericiguat (Verquvo) is unlikely to affect the pharmacokinetics of concurrently administered medications that are substrates of these enzymes or transporters.
In Vivo Assessment of Drug Interactions: No dose adjustment of Vericiguat (Verquvo) is recommended when co-administered with commonly prescribed medicinal products. There was no clinically relevant effect on vericiguat pharmacokinetics with coadministration of drugs increasing gastric pH (e.g. proton pump inhibitors, H2-receptor antagonists, antacids) in heart failure patients; or with coadministration of mefenamic acid, ketoconazole, rifampicin, digoxin, warfarin, aspirin, sildenafil, or the combination of sacubitril/valsartan in healthy subjects (see Figure 4 and Table 5). There was no clinically relevant effect on vericiguat pharmacokinetics with coadministration of atazanavir based on physiologically-based PK (PBPK) modeling (see Figure 4 and Table 5). Vericiguat also had no clinically relevant effect on the pharmacokinetics of midazolam, digoxin, warfarin, sildenafil, and the combination of sacubitril/valsartan when co-administered in healthy subjects (see Figure 5 and Table 6).
Effects of Other Drugs on the Pharmacokinetics of Vericiguat: The effects of co-administered drugs on the pharmacokinetics of vericiguat have been assessed in clinical drug-drug interaction studies (see Figure 4 and Table 5).
Drugs Increasing Gastric pH (e.g. Proton Pump Inhibitors, H2-receptor Antagonists, Antacids): Co-treatment with drugs that increase gastric pH, such as proton pump inhibitors, H2-receptor antagonists, or antacids, did not affect vericiguat exposure when vericiguat was taken as directed with food in heart failure patients [see Adults under Dosage & Administration].
Multi-pathway CYP and Transporter Inhibitor (Ketoconazole): Multiple-dose administration of ketoconazole 200 mg twice daily was not associated with a clinically relevant effect on the exposure of vericiguat 1.25 mg. The vericiguat mean AUC and mean Cmax following coadministration with ketoconazole were increased by approximately 12%.
UGT1A9 Inhibitor (Mefenamic Acid): A starting dose of mefenamic acid 500 mg followed by multiple-dose administration of 250 mg every 6 hours over 48 hours was not associated with a clinically relevant effect on the exposure of vericiguat 2.5 mg. The vericiguat mean AUC was increased by 20% and mean Cmax was decreased by 3%, following coadministration with mefenamic acid.
UGT1A1 Inhibitor (Atazanavir): Co-administration of atazanavir 400 mg once daily was not associated with a clinically relevant effect on the exposure of vericiguat 10 mg based on physiologically-based PK (PBPK) modeling. The predicted vericiguat mean AUC and mean Cmax were increased by 12% and 4%, respectively.
Broad Spectrum Inducer (Rifampicin): Multiple-dose administration of rifampicin 600 mg once daily for 8 days was not associated with a clinically relevant effect on the exposure of vericiguat 10 mg. The vericiguat mean AUC and mean Cmax following coadministration with rifampicin were decreased by 29% and 9%, respectively.
PDE-5 Inhibitor (Sildenafil): Single-dose administration of sildenafil 25, 50, and 100 mg was not associated with a clinically relevant effect on the exposure of multiple doses of vericiguat 10 mg once daily. The vericiguat mean AUC and mean Cmax following coadministration with sildenafil 25, 50, and 100 mg were changed by less than 4% and less than 9%, respectively. No dose-dependent effect on the pharmacokinetics of vericiguat was observed with the different sildenafil doses.
Effects of Vericiguat on the Pharmacokinetics of Other Drugs: The effects of vericiguat on the pharmacokinetics of coadministered drugs have been assessed in clinical drug-drug interaction studies (see Figure 5 and Table 6).
CYP3A Substrate (Midazolam): Multiple-dose administration of vericiguat 10 mg once daily for 4 days was not associated with a clinically relevant effect on the exposure of a single-dose of midazolam 7.5 mg. The midazolam mean AUC and mean Cmax following coadministration with vericiguat were decreased by 18% and 23%, respectively.
PDE-5 Inhibitor (Sildenafil): Multiple-dose administration of vericiguat 10 mg once daily was not associated with a clinically relevant effect on the exposure of a single-dose of sildenafil 25, 50, and 100 mg. The sildenafil 25, 50, and 100 mg mean AUC and mean Cmax following coadministration with vericiguat were increased by 13-22% and 14-20%, respectively.
Concomitant Use with Medicinal Products Commonly Prescribed to Heart Failure Patients: P-gp Substrate (Digoxin): Multiple-dose administration of digoxin 0.375 mg together with multiple doses of vericiguat 10 mg once daily was not associated with clinically relevant effects on the exposure (AUC and Ctrough) of digoxin. Multiple-dose administration of digoxin 0.375 mg together with a single dose of vericiguat 10 mg was not associated with clinically relevant effects on the exposure (AUC and Cmax) of vericiguat.
Anticoagulant (Warfarin): Single-dose administration of warfarin 25 mg together with multiple doses of vericiguat 10 mg once daily was not associated with clinically relevant effects on the exposure (AUC and Cmax) of either drug.
Antiplatelet Agent (Aspirin): Multiple-dose administration of aspirin 500 mg once daily together with a single-dose of vericiguat 15 mg was not associated with clinically relevant effects on the exposure (AUC and Cmax) of vericiguat.
Neprilysin Inhibitor/Angiotensin II Receptor Blocker (Combination of Sacubitril/Valsartan): Multiple-dose administration of the fixed dose combination of sacubitril 97 mg and valsartan 103 mg twice daily together with a single-dose of vericiguat 2.5 mg was not associated with clinically relevant effects on the exposure (AUC and Cmax) of vericiguat. The vericiguat mean AUC and mean Cmax following coadministration with sacubitril/valsartan were decreased by 7% and 9%, respectively. Multiple-dose administration of the fixed dose combination of sacubitril 97 mg and valsartan 103 mg twice daily together with multiple doses of vericiguat 2.5 mg once daily was not associated with clinically relevant effects on the exposure (AUC and Cmax) of sacubitril, LBQ657 (active metabolite of sacubitril), or valsartan. The sacubitril mean AUC and mean Cmax following coadministration with vericiguat were increased by 8% and 18%, respectively. The LBQ657 mean AUC and mean Cmax following coadministration with vericiguat were increased by 1% and 2%, respectively. The valsartan mean AUC and mean Cmax following coadministration with vericiguat were increased by 12% and 13%, respectively. (See Figures 4,5 and Tables 5,6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image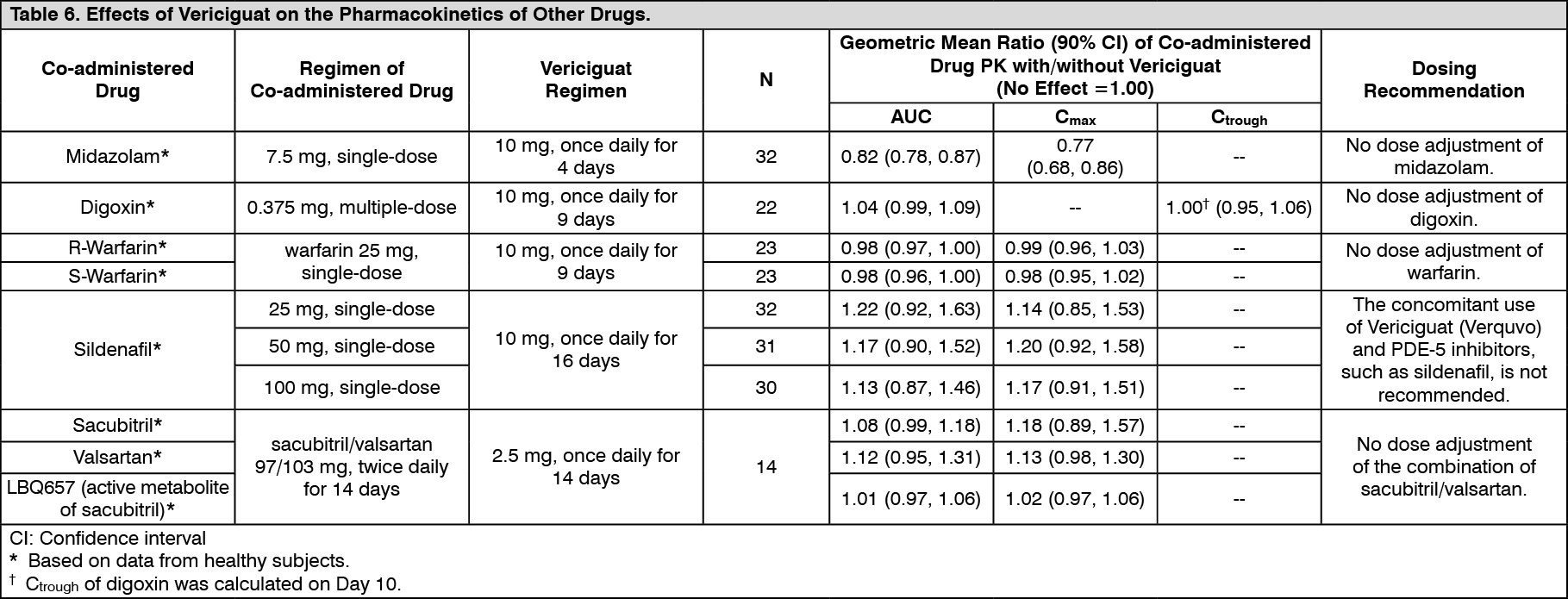 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacodynamic Interactions: Acetylsalicylic Acid (Aspirin): Administration of a single-dose of vericiguat 15 mg in healthy subjects did not alter the effect of acetylsalicylic acid 500 mg on bleeding time or platelet aggregation. Bleeding time or platelet aggregation did not change under treatment with vericiguat 15 mg alone.
Warfarin: Administration of multiple doses of vericiguat 10 mg once daily in healthy subjects did not alter the effect of a single-dose of warfarin 25 mg on prothrombin time and the activities of Factors II, VII, and X.
Combination of Sacubitril/Valsartan: Addition of multiple doses of vericiguat 2.5 mg to multiple doses of sacubitril/valsartan 97/103 mg in healthy subjects had no additional effect on seated blood pressure (BP) compared to administration of sacubitril/valsartan alone.
Sildenafil: Addition of single doses of sildenafil (25, 50, or 100 mg) to multiple doses of vericiguat 10 mg once daily in healthy subjects was associated with additional seated BP reduction of less than or equal to 5.4 mmHg (systolic/diastolic BP, MAP) compared to administration of vericiguat alone. No dose-dependent trend was observed with the different sildenafil doses [see Symptomatic Hypotension under Precautions].
Organic Nitrates: Co-administration of multiple doses of vericiguat increased to 10 mg once daily did not significantly alter the seated BP effects of short- and long-acting nitrates (nitroglycerin spray and isosorbide mononitrate [ISMN] modified release 60 mg) in patients with coronary artery disease. In patients with heart failure, concomitant use of short-acting nitrates was well tolerated. There is limited experience with concomitant use of vericiguat and long-acting nitrates in patients with heart failure [see Symptomatic Hypotension under Precautions].
Animal Toxicology: Acute Toxicity: No acute toxicity was observed in pivotal repeat-dose oral toxicity studies in rats up to 60 mg/kg/day and in dogs up to 25 mg/kg/day (approximately 75 or 12 times the human exposure [unbound AUC] at the maximum recommended human dose [MRHD] of 10 mg/day).
Chronic Toxicity: Repeat-dose oral toxicity studies were conducted in rats and dogs for up to 26 and 39 weeks, respectively. In the chronic toxicity studies, no adverse signs of toxicity were observed up to exposures equal to approximately 50 (rat) or 8 (dog) times the human exposure (unbound AUC) at the MRHD of 10 mg/day.
The toxicological profile was characterized by effects secondary to exaggerated pharmacodynamics. Secondary to smooth muscle relaxation hemodynamic and gastrointestinal effects were noted in all species investigated. In adolescent rapidly-growing rats, reversible bone effects consisting of hypertrophy of growth plate and hyperostosis and remodeling of metaphyseal and diaphyseal bone were seen that were mediated by a mode of action-related intracellular cGMP increase. These effects were not observed after chronic administration of vericiguat to adult rats up to exposures of approximately 50 times the human exposure at the MRHD. In addition, no comparable findings were seen with dogs which were almost full-grown at start of treatment up to exposures of 15 times the human exposure at the MRHD.
Carcinogenesis: Carcinogenicity was evaluated in 2-year studies conducted in CD1 mice and Wistar rats.
Vericiguat did not show a carcinogenic effect in mice dosed up to 150 mg/kg/day (males) or up to 250 mg/kg/day (females). These doses were associated with exposures 149 (males) or 286 (females) times the human exposure (unbound AUC) at the MRHD of 10 mg/day.
In the carcinogenicity study in rats, no vericiguat-related tumor or hyperplastic findings were seen up to exposures of 12 times the human exposure at the MRHD. A non-statistical numerical increase of benign pheochromocytomas and Leydig cell tumors as well as respective hyperplasias were observed in males after administration of the high dose of 20 mg/kg/day leading to exposure of 41 times the human exposure at the MRHD. This is considered a consequence of a compensatory and recurrent activation of the renin angiotensin aldosterone and the adrenergic system due to a marked daily decrease in blood pressure over 2 years. Based on the known sensitivity of rats to develop these two tumor types in contrast to humans and a documented pharmacological-based mechanism (seen also with other antihypertensive drugs) at supratherapeutic doses as well as adequate safety margins this is considered not relevant for patients.
Non-clinical data revealed no carcinogenic risk for humans at clinical doses.
Mutagenesis: Vericiguat was not genotoxic in the in vitro microbial mutagenicity (Ames) assay, the in vitro mouse lymphoma assay, and the in vivo rat and mouse micronucleus assay.
Reproduction: In a 4-week repeat dose fertility and early embryonic development study in male and female rats, vericiguat when administered orally at doses of 5, 15 or 50 mg/kg/day had no effects on fertility or reproductive performance at up to the highest dose tested of 50 mg/kg/day (66 times the human exposure at the MRHD of 10 mg/day, unbound AUC).
Development: Reproductive toxicity studies with vericiguat showed no evidence of developmental toxicity (rats, rabbits) or effects on pre/postnatal development (rats).
In a prenatal developmental toxicity study in rats, vericiguat was administered orally to pregnant rats during the period of organogenesis from gestation days (GD) 6 to 17 at doses of 5, 15 or 50 mg/kg/day. No developmental toxicity was observed up to the highest dose (75 times the human exposure at the MRHD, unbound AUC). Exaggerated pharmacodynamic-mediated maternal toxicity (decreased body weight gain and food consumption) was observed at ≥15 mg/kg/day (≥21 times the human exposure at the MRHD).
There was no maternal toxicity at 5 mg/kg/day (9 times the human exposure at the MRHD).
In a prenatal developmental toxicity study in rabbits, vericiguat was administered orally to pregnant rabbits during the period of organogenesis from GD 6 to 20 at doses of 0.75, 2.50 or 7.50 mg/kg/day. No developmental toxicity was observed up to the highest dose tested (27 times the human exposure at the MRHD). Exaggerated pharmacodynamic-mediated maternal toxicity (decreased food consumption and body weight loss) resulting in late spontaneous abortions and resorptions was noted at ≥2.50 mg/kg/day (≥6 times the human exposure at the MRHD). There was no maternal toxicity or abortions/resorptions in rabbits at an exposure equivalent to the human exposure at the MRHD.
In a pre-postnatal development study in rats, vericiguat was administered orally at doses of 7.5, 15 or 30 mg/kg/day from GD 6 through lactation day 21. Exaggerated pharmacodynamic-mediated maternal toxicity (decreases in food consumption and body weight gain) was observed at all dose levels (≥9 times at the MRHD) and resulted in decreased pup body weight gain at ≥15 mg/kg/day (≥21 times at the MRHD) and pup mortality at 30 mg/kg/day (49 times at the MHRD).
[14C]-vericiguat was administered orally to pregnant rats at a dose of 3 mg/kg. Vericiguat-related material was transferred across the placenta, with fetal plasma concentrations of approximately 67% maternal concentrations on GD 19.
[14C]-vericiguat was administered intravenously to lactating rats at a dose of 1 mg/kg. Vericiguat-related material was excreted into milk at concentrations approximately 12% maternal plasma concentrations on LD 8.