Pharmacologic Category: Antibacterial.
Pharmacology: Pharmacodynamics: Mechanism of action: Tigecycline, a glycylcycline antibiotic, inhibits protein translation in bacteria by binding to the 30S ribosomal subunit and blocking entry of amino-acyl tRNA molecules into the A site of the ribosome. This prevents incorporation of amino acid residues into elongating peptide chains. Tigecycline carries a glycylamido moiety attached to the 9-position of minocycline. The substitution pattern is not present in any naturally occurring or semisynthetic tetracycline and imparts certain microbiologic properties that transcend any known tetracycline-derivative
in vitro or
in vivo activity. In addition, tigecycline is able to overcome the two major tetracycline resistance mechanisms, ribosomal protection and efflux. Accordingly, tigecycline has demonstrated
in vitro and
in vivo activity against a broad spectrum of bacterial pathogens. There has been no cross resistance observed between tigecycline and other antibiotics. In
in vitro studies, no antagonism has been observed between tigecycline and other commonly used antibiotics. In general, tigecycline is considered bacteriostatic. At 4 times the minimum inhibitory concentration (MIC), a 2-log reduction in colony counts was observed with tigecycline against
Enterococcus spp.,
Staphylococcus aureus, and
Escherichia coli. However, tigecycline has shown some bactericidal activity, and a 3-log reduction was observed against
Neisseria gonorrhoeae. Tigecycline has also demonstrated bactericidal activity against common respiratory strains of
Streptococcus pneumoniae,
Haemophilus influenzae, and
Legionella pneumophila.
Susceptibility Test Methods: Dilution Techniques: Quantitative methods are used to determine antimicrobial MICs. These MICs provide estimates of the susceptibility of bacteria to antimicrobial compounds. The MICs should be determined using a standardized procedure based on dilution methods (broth, agar, or microdilution) or equivalent using standardized inoculum and concentrations of tigecycline. For broth dilution tests for aerobic organisms, MICs must be determined in testing medium that is fresh (<12 hours old). The MIC values should be interpreted according to the criteria provided in Table 1.
Diffusion Techniques:
Quantitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. The standardized procedure requires the use of standardized inoculum concentrations. This procedure uses paper disks impregnated with 15 μg tigecycline to test the susceptibility of microorganisms to tigecycline. Interpretation involves correlation of the diameter obtained in the disk test with the MIC for tigecycline. Reports from the laboratory providing results of the standard single-disk susceptibility test with a 15 μg tigecycline disk should be interpreted according to the criteria in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
A report of "Susceptible" indicates that the pathogen is likely to be inhibited if the antimicrobial compound reaches the concentrations usually achievable. A report of "Intermediate" indicates that the result should be considered equivocal, and if the microorganism is not fully susceptible to alternative, clinically feasible drugs, the test should be repeated. This category implies possible clinical applicability in body sites where the drug is physiologically concentrated or in situations where high dosage of drug can be used. This category also provides a buffer zone that prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of "Resistant" indicates that the pathogen is not likely to be inhibited if the antimicrobial compound reaches the concentrations usually achievable; other therapy should be selected.
Quality Control: As with other susceptibility techniques, the use of laboratory control microorganisms is required to control the technical aspects of the laboratory standardized procedures. Standard tigecycline powder should provide the MIC values provided in Table 2. For the diffusion technique using the 15 μg tigecycline disk, laboratories should use the criteria provided in Table 2 to test quality control strains. (See Table 2.)
Click on icon to see table/diagram/image
The prevalence of acquired resistance may vary geographically and with time for selected species, and local information on resistance is desirable, particularly when treating severe infections. The information as follows provides only approximate guidance on the probability as to whether the microorganism will be susceptible to tigecycline or not:
Susceptible: Gram-positive aerobes:
Enterococcus avium,
Enterococcus casseliflavus,
Enterococcus faecalis* (includes vancomycin-susceptible strains),
Enterococcus faecalis (includes vancomycin-resistant strains),
Enterococcus faecium (includes vancomycin-susceptible and -resistant strains),
Enterococcus gallinarum,
Listeria monocytogenes,
Staphylococcus aureus* (includes methicillin-susceptible and -resistant strains, including isolates that bear molecular and virulence markers commonly associated with community-acquired MRSA including the SCCmec type IV element and the pvl gene),
Staphylococcus epidermidis (includes methicillin-susceptible and -resistant strains),
Staphylococcus haemolyticus,
Streptococcus agalactiae*,
Streptococcus anginosus* (includes
S. anginosus,
S. intermedius,
S. constellatus),
Streptococcus pyogenes*,
Streptococcus pneumoniae* (penicillin-susceptible isolates),
Streptococcus pneumoniae (penicillin-resistant isolates), Viridans group streptococci.
Gram-negative aerobes:
Acinetobacter calcoaceticus/
baumannii complex,
Aeromonas hydrophila,
Citrobacter freundii*,
Citrobacter koseri,
Enterobacter aerogenes,
Enterobacter cloacae*,
Escherichia coli* (including extended spectrum beta lactamase-producing strains),
Haemophilus influenzae*,
Haemophilus parainfluenzae,
Klebsiella oxytoca*,
Klebsiella pneumoniae* (including extended spectrum beta lactamase-producing strains),
Klebsiella pneumoniae (including AmpC producing strains),
Legionella pneumophila*,
Moraxella catarrhalis*,
Neisseria gonorrhoeae,
Neisseria meningitidis,
Pasteurella multocida,
Salmonella enterica ser.
Enteritidis,
Salmonella enterica ser. Paratyphi,
Salmonella entericaser. Typhi,
Salmonella enterica ser. Typhimurium,
Serratia marcescens,
Shigella boydii,
Shigella dysenteriae,
Shigella flexneri,
Shigella sonnei,
Stenotrophomonas maltophilia.
Anaerobic bacteria:
Bacteroides fragilis*,
Bacteroides distasonis,
Bacteroides ovatus,
Bacteroides thetaiotaomicron*,
Bacteroides uniformis*,
Bacteroides vulgatus*,
Clostridium difficile,
Clostridium perfringens*,
Peptostreptococcus spp.,
Peptostreptococcus micros*,
Porphyromonas spp.,
Prevotella spp.
Atypical bacteria:
Chlamydia pneumoniae*,
Mycobacterium abscessus,
Mycobacterium chelonae,
Mycobacterium fortuitum,
Mycoplasma pneumoniae*.
*Clinical efficacy has been demonstrated for susceptible isolates in the approved clinical indications.
Resistant: Gram-negative aerobes:
Pseudomonas aeruginosa.
Anaerobic bacteria: No naturally-occurring species have been found to be inherently resistant to tigecycline.
Resistance: There has been no cross-resistance observed between tigecycline and other antibiotics.
Tigecycline is able to overcome the two major tetracycline resistance mechanisms, ribosomal protection and efflux.
In
in vitro studies, no antagonism has been observed between tigecycline and any other commonly used antibiotic class.
Clinical trial data on efficacy: Complicated Skin and Skin Structure Infections (cSSSI): Tigecycline was evaluated in adults for the treatment of cSSSI in two randomized, double-blind, active-controlled, multinational, multicenter studies. These studies compared tigecycline (100 mg IV initial dose followed by 50 mg every 12 hours) with vancomycin (1 g IV every 12 hours)/aztreonam (2 g IV every 12 hours) for 5 to 14 days. Subjects with complicated deep soft-tissue infections, including wound infections and cellulitis (≥10 cm, requiring surgery/drainage or with complicated underlying disease), major abscesses, infected ulcers, and burns were enrolled in the studies. The primary efficacy endpoint was the clinical response at the test of cure (TOC) visit in the co-primary populations of the clinically evaluable (CE) and clinical modified intent-to-treat (c-mITT) subjects. (See Table 3.)
Click on icon to see table/diagram/image
Clinical cure rates at TOC by pathogen in microbiologically evaluable (ME) subjects with cSSSI are presented in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
Tigecycline did not meet non-inferiority criteria in comparison with ertapenem in a study of subjects with diabetic foot infection (See Table 5). This was a randomized, double-blind, multinational, multicenter trial comparing tigecycline (150 mg every 24 hours) with ertapenem (1 g every 24 hours, with or without vancomycin) for up to 28 days. The primary efficacy endpoint was the clinical response at the TOC assessment in the co-primary CE and c-mITT populations. The non-inferiority margin was -10% for the difference in cure rates between the 2 treatments. (See Table 5.)
Click on icon to see table/diagram/image
Complicated Intra-abdominal Infections (cIAI): Tigecycline was evaluated in adults for the treatment of cIAI in two randomized, double-blind, active-controlled, multinational, multicenter studies. These studies compared tigecycline (100 mg IV initial dose followed by 50 mg every 12 hours) with imipenem/cilastatin (500 mg IV every 6 hours) for 5 to 14 days. Subjects with complicated diagnoses including appendicitis, cholecystitis, diverticulitis, gastric/duodenal perforation, intra-abdominal abscess, perforation of the intestine, and peritonitis were enrolled in the studies. The primary efficacy endpoint was the clinical response at the TOC visit for the co-primary populations of the ME and the microbiologic modified intent-to-treat (m-mITT) subjects. (See Table 6.)
Click on icon to see table/diagram/image
Clinical cure rates at TOC by pathogen in ME subjects with cIAI are presented in Table 7. (See Table 7.)
Click on icon to see table/diagram/image
Community-Acquired Pneumonia (CAP): Tigecycline was evaluated in adults for the treatment of CAP in two randomized, double-blind, active-controlled, multinational, multicenter studies (Studies 308 and 313). These studies compared tigecycline (100 mg IV initial dose followed by 50 mg every 12 hours) with levofloxacin (500 mg IV every 12 or 24 hours). In one study (Study 308), after at least 3 days of IV therapy, a switch to oral levofloxacin (500 mg daily) was permitted for both treatment arms. Total therapy was 7 to 14 days. Subjects with CAP who required hospitalization and IV therapy were enrolled in the studies. The primary efficacy endpoint was the clinical response at the TOC visit in the co-primary populations of the CE and c-mITT subjects. See Table 8. Clinical cure rates at TOC by pathogen in the ME subjects are presented in Table 9. (See Tables 8 and 9.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Methicillin-resistant
Staphylococcus aureus (MRSA) and Vancomycin-resistant
Enterococcus (VRE) spp.: Tigecycline was evaluated in adults for the treatment of various serious infections (cIAI, cSSSI, and other infections) due to VRE and MRSA in Study 307.
Study 307 was a randomized, double-blind, active-controlled, multinational, multicenter study evaluating tigecycline (100 mg IV initial dose followed by 50 mg every 12 hours) and vancomycin (1 g IV every 12 hours) for the treatment of infections due to MRSA and evaluating tigecycline (100 mg IV initial dose followed by 50 mg every 12 hours) and linezolid (600 mg IV every 12 hours) for the treatment of infections due to VRE for 7 to 28 days. Subjects with cIAI, cSSSI, and other infections were enrolled in this study. The primary efficacy endpoint was the clinical response at the TOC visit for the co-primary populations of the ME and the m-mITT subjects. For clinical cure rates, see Table 10 for MRSA and Table 11 for VRE. (See Tables 10 and 11.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Resistant Gram-negative Pathogens: Tigecycline was evaluated in adults for the treatment of various serious infections (cIAI, cSSSI, CAP, and other infections) due to resistant gram-negative pathogens in Study 309.
Study 309 was an open-label, multinational, multicenter study evaluating tigecycline (100 mg IV initial dose followed by 50 mg every 12 hours) for the treatment of infections due to resistant gram-negative pathogens for 7 to 28 days. Subjects with cIAI, cSSSI, CAP, and other infections were enrolled in this study. The primary efficacy endpoint was the clinical response at the TOC visit for the co-primary populations of the ME and the m-mITT subjects. (See Table 12.)
Click on icon to see table/diagram/image
Rapidly Growing Mycobacterial Infections: In uncontrolled clinical studies and compassionate-use experience from 8 countries, 52 subjects with rapidly-growing mycobacterial infections (most frequently
M. abscessus lung disease) were treated with tigecycline, along with other antibiotics. The mean and median durations of treatment were approximately 5½ months and 3 months, respectively (range: 3 days to approximately 3½ years). Approximately half of the subjects achieved clinical improvement (i.e., improvement in signs and symptoms of lung disease, or healing of wound, skin lesions, or nodules in disseminated disease). Approximately half of the subjects required dose reductions or discontinued treatment due to nausea, vomiting, or anorexia.
Pediatric population: In an open-label, ascending multiple-dose study, 39 children aged 8 to 11 years with cIAI or cSSSI were administered tigecycline (0.75, 1, or 1.25 mg/kg). All patients received IV tigecycline for a minimum of 3 consecutive days to a maximum of 14 consecutive days, with the option to be switched to an oral antibiotic on or after day 4.
Clinical cure was assessed between 10 and 21 days after the administration of the last dose of treatment. The summary of clinical response in the modified intent-to-treat (mITT) population results is shown in following table. (See Table 13.)
Click on icon to see table/diagram/image
Efficacy data shown previously should be viewed with caution as concomitant antibiotics were allowed in this study. In addition, the small number of patients should also be taken into consideration.
Cardiac Electrophysiology: No significant effect of a single intravenous dose of tigecycline 50 mg or 200 mg on QTc interval was detected in a randomized, placebo- and active-controlled four-arm crossover thorough QTc study of 46 healthy subjects.
Pharmacokinetics: The mean pharmacokinetic parameters of tigecycline for the recommended dosage regimen after single and multiple intravenous doses are summarized in Table 14.
Intravenous infusions of tigecycline should be administered over approximately 30 to 60 minutes. (See Table 14.)
Click on icon to see table/diagram/image
Absorption: Tigecycline is administered intravenously, and therefore has 100% bioavailability.
Distribution: The
in vitro plasma protein binding of tigecycline ranges from approximately 71% to 89% at concentrations observed in clinical studies (0.1 to 1.0 μg/mL). Animal and human pharmacokinetic studies have demonstrated that tigecycline readily distributes to tissues. In rats receiving a single or multiple doses of C-tigecycline, radioactivity was well distributed to most tissues, with the highest overall exposure observed in bone, bone marrow, thyroid gland, kidney, spleen, and salivary gland. In humans, the steady-state volume of distribution of tigecycline averaged 500 to 700 L (7 to 9 L/kg), indicating that tigecycline is extensively distributed beyond the plasma volume and into the tissues of humans.
Two studies examined the steady-state pharmacokinetic profile of tigecycline in specific tissues or fluids of healthy subjects receiving tigecycline 100 mg followed by 50 mg every 12 hours. In a bronchoalveolar lavage study, the tigecycline AUC
0-12h (134 μg•h/mL) in alveolar cells was approximately 77.5-fold higher than the AUC
0-12h in the serum of these subjects, and the AUC
0-12h (2.28 μg•h/mL) in epithelial lining fluid was approximately 32% higher than the AUC
0-12h in serum. In a skin blister study, the AUC
0-12h (1.61 μg•hr/mL) of tigecycline in skin blister fluid was approximately 26% lower than the AUC
0-12h in the serum of these subjects.
In a single-dose study, tigecycline 100 mg was administered to subjects prior to undergoing elective surgery or medical procedure for tissue extraction. Tissue concentrations at 4 hours after tigecycline administration were measured in the following tissue and fluid samples: gallbladder, lung, colon, synovial fluid, and bone. Tigecycline attained higher concentrations in tissues versus serum in gallbladder (38-fold, n=6), lung (3.7-fold, n=5), and colon (2.3-fold, n=6). The concentration of tigecycline in these tissues after multiple doses has not been studied.
Metabolism: Tigecycline is not extensively metabolized.
In vitro studies with tigecycline using human liver microsomes, liver slices, and hepatocytes led to the formation of only trace amounts of metabolites. In healthy male volunteers receiving
14C-tigecycline, tigecycline was the primary
14C-labeled material recovered in urine and feces, but a glucuronide, an N-acetyl metabolite and a tigecycline epimer (each at no more than 10% of the administered dose) were also present.
Elimination: The recovery of total radioactivity in feces and urine following administration of
14C-tigecycline indicates that 59% of the dose is eliminated by biliary/fecal excretion, and 33% is excreted in urine. Overall, the primary route of elimination for tigecycline is biliary excretion of unchanged tigecycline. Glucuronidation and renal excretion of unchanged tigecycline are secondary routes.
Tigecycline is a substrate of P-gp based on an
in vitro study using a cell line overexpressing P-gp. The potential contribution of P-gp-mediated transport to the
in vivo disposition of tigecycline is not known.
Special populations: Hepatic insufficiency: In a study comparing 10 subjects with mild hepatic impairment (Child Pugh A), 10 subjects with moderate hepatic impairment (Child Pugh B), and five subjects with severe hepatic impairment (Child Pugh C) to 23 age- and weight-matched healthy control subjects, the single-dose pharmacokinetic disposition of tigecycline was not altered in subjects with mild hepatic impairment. However, systemic clearance of tigecycline was reduced by 25%, and the half-life of tigecycline was prolonged by 23% in subjects with moderate hepatic impairment (Child Pugh B). In addition, systemic clearance of tigecycline was reduced by 55%, and the half-life of tigecycline was prolonged by 43% in subjects with severe hepatic impairment (Child Pugh C).
Based on the pharmacokinetic profile of tigecycline, no dosage adjustment is warranted in subjects (including pediatrics) with mild to moderate hepatic impairment (Child Pugh A and Child Pugh B). However, in subjects with severe hepatic impairment (Child Pugh C), the dose of tigecycline should be reduced by 50%. Adult dose should be altered to 100 mg followed by 25 mg every 12 hours. Subjects with severe hepatic impairment (Child Pugh C) should be treated with caution and monitored for treatment response (see Dosage & Administration).
Renal insufficiency: A single-dose study compared 6 subjects with severe renal impairment (creatinine clearance ClCr ≤30 mL/min), 4 end stage renal disease subjects receiving tigecycline 2 hours before hemodialysis, 4 end stage renal disease subjects receiving tigecycline after hemodialysis, and 6 healthy control subjects. The pharmacokinetic profile of tigecycline was not altered in any of the renally-impaired subject groups, nor was tigecycline removed by hemodialysis. No dosage adjustment of tigecycline is necessary in subjects with renal impairment or in subjects undergoing hemodialysis (see Dosage & Administration).
Elderly: No overall differences in pharmacokinetics were observed between healthy elderly subjects (n=15, age 65-75; n=13, age 75, and younger subjects (n=18) receiving a single, 100 mg dose of tigecycline. Therefore, no dosage adjustment is necessary based on age.
Children: Tigecycline pharmacokinetics were investigated in two studies. The first study enrolled children aged 8-16 years (n=24) who received single doses of tigecycline (0.5, 1, or 2 mg/kg, up to a maximum dose of 50 mg, 100 mg, and 150 mg, respectively) administered intravenously over 30 minutes. The second study was performed in children aged 8 to 11 years who received multiple doses of tigecycline (0.75, 1, or 1.25 mg/kg up to a maximum dose of 50 mg) every 12 hours administered intravenously over 30 minutes. No loading dose was administered in these studies. Pharmacokinetic parameters are summarised in the table as follows. (See Table 15.)
Click on icon to see table/diagram/image
The target AUC
0-12h in adults after the recommended dose of 100 mg loading and 50 mg every 12 hours, was approximately 2500 ng•h/mL.
Population PK analysis of both studies identified body weight as a covariate of tigecycline clearance in children aged 8 years and older. A dosing regimen of 1.2 mg/kg of tigecycline every 12 hours (to a maximum dose of 50 mg every 12 hours) for children aged 8 to <12 years, and of 50 mg every 12 hours for adolescents aged 12 to <18 years would likely result in exposures comparable to those observed in adults treated with the approved dosing regimen.
Gender: In a pooled analysis of 38 women and 298 men participating in clinical pharmacology studies, there was no significant difference in the mean (±SD) tigecycline clearance between women (20.7±6.5 L/h) and men (22.8±8.7 L/h). Therefore, no dosage adjustment is necessary based on gender.
Race: In a pooled analysis of 73 Asian subjects, 53 Black subjects, 15 Hispanic subjects, 190 White subjects, and 3 subjects classified as "other" participating in clinical pharmacology studies, there was no significant difference in the mean (±SD) tigecycline clearance among the Asian subjects (28.8±8.8 L/h), Black subjects (23.0±7.8 L/h), Hispanic subjects (24.3±6.5 L/h), White subjects (22.1±8.9 L/h), and "other" subjects (25.0±4.8 L/h). Therefore, no dosage adjustment is necessary based on race.
Toxicology: Preclinical Safety Data: Carcinogenicity: Lifetime studies in animals have not been performed to evaluate the carcinogenic potential of tigecycline.
Mutagenicity: No mutagenic or clastogenic potential was found in a battery of tests, including an
in vitro chromosome aberration assay in Chinese hamster ovary (CHO) cells,
in vitro forward mutation assay in CHO cells (HGRPT locus),
in vitro forward mutation assays in mouse lymphoma cells, and
in vivo micronucleus assay.
Impairment of fertility: Tigecycline did not affect mating or fertility in rats at exposures up to 4.7 times the human daily dose based on AUC. In female rats, there were no compound-related effects on ovaries or estrus cycles at exposures up to 4.7 times the human daily dose based on AUC.
Other: Decreased erythrocytes, reticulocytes, leukocytes and platelets, in association with bone marrow hypocellularity, have been seen with tigecycline at exposures of 8.1 times and 9.8 times the human daily dose based on AUC in rats and dogs, respectively. These alterations were shown to be reversible after two weeks of dosing.
Bolus intravenous administration of tigecycline has been associated with a histamine response in preclinical studies. These effects were observed at exposures of 14.3 and 2.8 times the human daily dose based on the AUC in rats and dogs, respectively.
No evidence of photosensitivity was observed in rats following administration of tigecycline.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image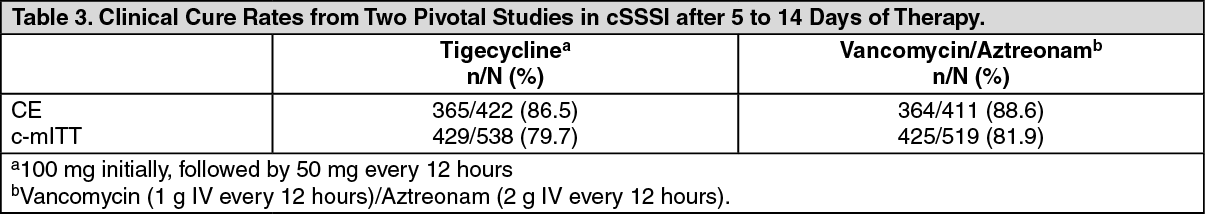 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image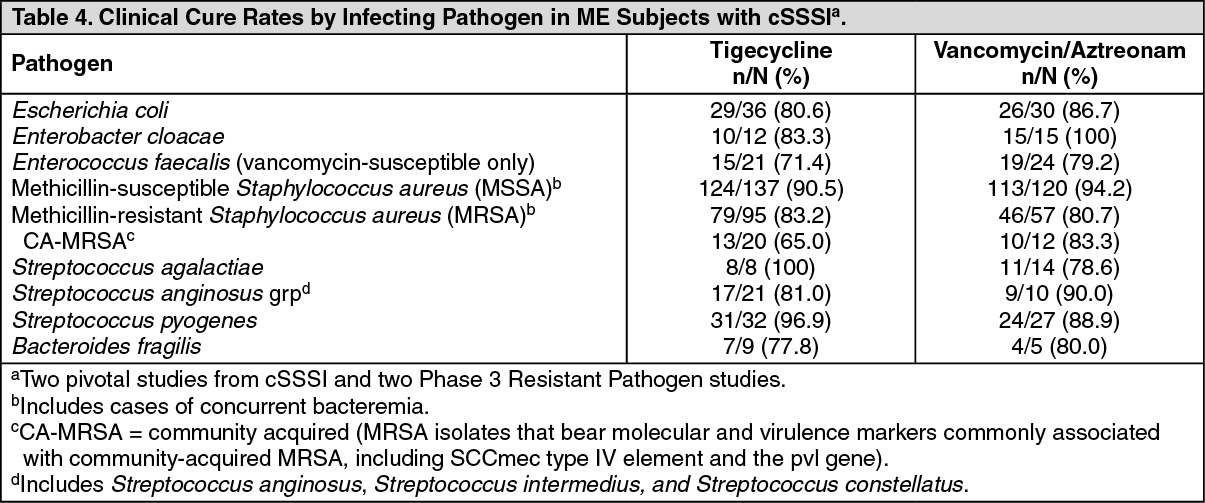 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image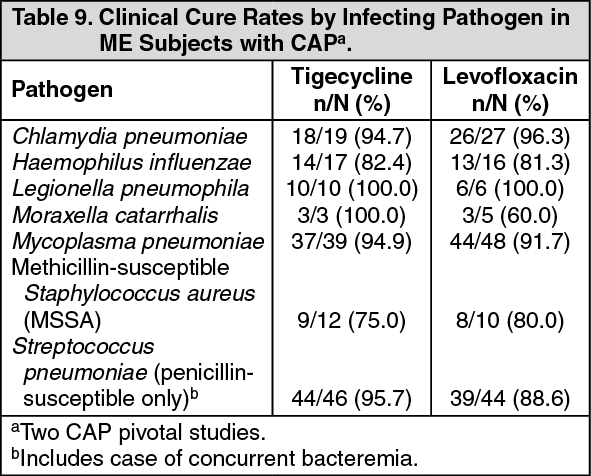 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image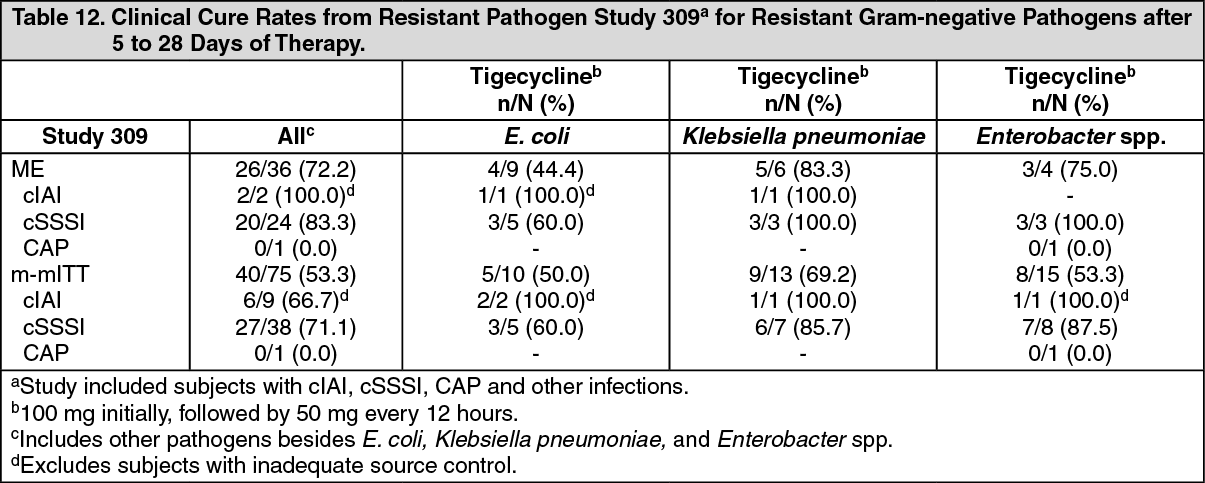 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image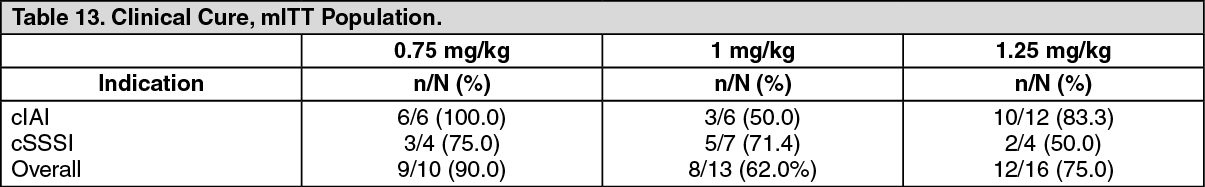 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image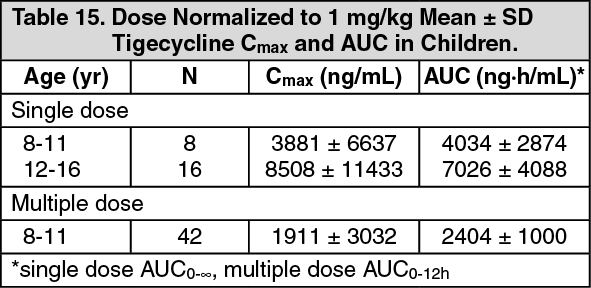 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
