Pharmacology: Mechanism of Action: Efavirenz, lamivudine and tenofovir disoproxil fumarate tablets are a fixed dose combination of antiviral drugs efavirenz, lamivudine and tenofovir disoproxil fumarate.
Efavirenz: Efavirenz is an NNRTI of HIV-1. Efavirenz activity is mediated predominantly by noncompetitive inhibition of HIV-1 reverse transcriptase. HIV-2 reverse transcriptase and human cellular DNA polymerases α, 3, y, and ö are not inhibited by efavirenz.
Lamivudine: Lamivudine is a synthetic nucleoside analogue. Intracellularly, lamivudine is phosphorylated to its active 5′-triphosphate metabolite, lamivudine triphosphate (3TC-TP). The principal mode of action of 3TC-TP is inhibition of HIV-1 reverse transcriptase (RT) via DNA chain termination after incorporation of the nucleotide analogue.
Tenofovir Disoproxil Fumarate: Tenofovir disoproxil fumarate is an acyclic nucleoside phosphonate diester analog of adenosine monophosphate. Tenofovir disoproxil fumarate requires initial diester hydrolysis for conversion to tenofovir and subsequent phosphorylations by cellular enzymes to form tenofovir diphosphate (TFV-DP), an obligate chain terminator. Tenofovir diphosphate inhibits the activity of HIV-1 reverse transcriptase (RT) and HBV RT by competing with the natural substrate deoxyadenosine 5'-triphosphate and, after incorporation into DNA, by DNA chain termination. Tenofovir diphosphate is a weak inhibitor of mammalian DNA polymerases α, 3, and mitochondrial DNA polymerase y.
Pharmacodynamics: Cardiac Electrophysiology: The effect of efavirenz on the QTc interval was evaluated in an open-label, positive and placebo controlled, fixed single sequence 3-period, 3-treatment crossover QT study in 58 healthy subjects enriched for CYP2B6 polymorphisms. The mean Cmax of efavirenz in subjects with CYP2B6 *6/*6 genotype following the administration of 600 mg daily dose for 14 days was 2.25-fold the mean Cmax observed in subjects with CYP2B6 *1/*1 genotype. A positive relationship between efavirenz concentration and QTc prolongation was observed. Based on the concentration-QTc relationship, the mean QTc prolongation and its upper bound 90% confidence interval are 8.7 ms and 11.3 ms in subjects with CYP2B6*6/*6 genotype following the administration of 600 mg daily dose for 14 days (see Precautions).
Antiviral Activity: Efavirenz: The concentration of efavirenz inhibiting replication of wild-type laboratory adapted strains and clinical isolates in cell culture by 90-95% (EC90-95) ranged from 1.7 to 25 nM in lymphoblastoid cell lines, peripheral blood mononuclear cells (PBMCs), and macrophage/monocyte cultures. Efavirenz demonstrated antiviral activity against clade B and most non-clade B isolates (subtypes A, AE, AG, C, D, F, G, J, N), but had reduced antiviral activity against group O viruses. Efavirenz demonstrated additive antiviral activity without cytotoxicity against HIV-1 in cell culture when combined with the NNRTIs delavirdine and nevirapine, NRTIs (abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, zalcitabine, zidovudine), PIs (amprenavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir), and the fusion inhibitor enfuvirtide. Efavirenz demonstrated additive to antagonistic antiviral activity in cell culture with atazanavir. Efavirenz was not antagonistic with adefovir, used for the treatment of hepatitis B virus infection, or ribavirin, used in combination with interferon for the treatment of hepatitis C virus infection.
Lamivudine: The antiviral activity of lamivudine against HIV-1 was assessed in a number of cell lines including monocytes and fresh human peripheral blood lymphocytes (PBMCs) using standard susceptibility assays. EC50 values were in the range of 0.003 to 15 microM (1 microM = 0.23 mcg per mL). The median EC50 values of lamivudine were 60 nM (range: 20 to 70 nM), 35 nM (range: 30 to 40 nM), 30 nM (range: 20 to 90 nM), 20 nM (range: 3 to 40 nM), 30 nM (range: 1 to 60 nM), 30 nM (range: 20 to 70 nM), 30 nM (range: 3 to 70 nM), and 30 nM (range: 20 to 90 nM) against HIV-1 clades A-G and group O viruses (n = 3 except n = 2 for clade B) respectively. The EC50 values against HIV-2 isolates (n = 4) ranged from 0.003 to 0.120 microM in PBMCs. Lamivudine was not antagonistic to all tested anti-HIV agents. Ribavirin (50 microM) used in the treatment of chronic HCV infection decreased the anti-HIV-1 activity of lamivudine by 3.5-fold in MT-4 cells.
Tenofovir Disoproxil Fumarate: The antiviral activity of tenofovir against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, primary monocyte/macrophage cells and peripheral blood lymphocytes. The EC50 (50% effective concentration) values for tenofovir were in the range of 0.04 jtM to 8.5 jtM. In drug combination studies, tenofovir was not antagonistic with HIV-1 NRTIs (abacavir, didanosine, lamivudine, stavudine, zidovudine), NNRTIs (efavirenz, nevirapine), and protease inhibitors (amprenavir, indinavir, nelfinavir, ritonavir, saquinavir). Tenofovir displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, G, and O (EC50 values ranged from 0.5 jtM to 2.2 jtM) and strain-specific activity against HIV-2 (EC50 values ranged from 1.6 jtM to 5.5 jtM).
Resistance: Efavirenz: In cell culture: In cell culture, HIV-1 isolates with reduced susceptibility to efavirenz (>380-fold increase in EC90 value) emerged rapidly in the presence of drug. Genotypic characterization of these viruses identified single amino acid substitutions L100I or V179D, double substitutions L100I/V108I, and triple substitutions L100I/V179D/Y181C in reverse transcriptase.
Clinical studies: Clinical isolates with reduced susceptibility in cell culture to efavirenz have been obtained. One or more substitutions at amino acid positions 98, 100, 101, 103, 106, 108, 188, 190, 225, and 227 in reverse transcriptase were observed in patients failing treatment with efavirenz in combination with indinavir, or with zidovudine plus lamivudine. The K103N substitution was the most frequently observed. Long-term resistance surveillance (average 52 weeks, range 4-106 weeks) analyzed 28 matching baseline and virologic failure isolates. Sixty-one percent (17/28) of these failure isolates had decreased efavirenz susceptibility in cell culture with a median 88-fold change in efavirenz susceptibility (EC50 value) from reference. The most frequent NNRTI substitution to develop in these patient isolates was K103N (54%). Other NNRTI substitutions that developed included L100I (7%), K101E/Q/R (14%), V108I (11%), G190S/T/A (7%), P225H (18%), and M230I/L (11%).
Lamivudine: Lamivudine-resistant variants of HIV-1 have been selected in cell culture. Genotypic analysis showed that the resistance was due to a specific amino acid substitution in the HIV-1 reverse transcriptase at codon 184 changing the methionine to either valine or isoleucine (M184V/I).
HIV-1 strains resistant to both lamivudine and zidovudine have been isolated from subjects. Susceptibility of clinical isolates to lamivudine and zidovudine was monitored in controlled clinical trials. In subjects receiving lamivudine monotherapy or combination therapy with lamivudine plus zidovudine, HIV-1 isolates from most subjects became phenotypically and genotypically resistant to lamivudine within 12 weeks.
Genotypic and Phenotypic Analysis of On-Therapy HIV-1 Isolates from Subjects with Virologic Failure: Trial EPV20001: Fifty-three of 554 (10%) subjects enrolled in EPV20001 were identified as virological failures (plasma HIV-1 RNA level greater than or equal to 400 copies per mL) by Week 48. Twenty-eight subjects were randomized to the lamivudine once-daily treatment group and 25 to the lamivudine twice-daily treatment group. The median baseline plasma HIV-1 RNA levels of subjects in the lamivudine once-daily group and lamivudine twice-daily group were 4.9 log10 copies per mL and 4.6 log10 copies per mL, respectively.
Genotypic analysis of on-therapy isolates from 22 subjects identified as virologic failures in the lamivudine once-daily group showed that isolates from 8 of 22 subjects contained a treatment-emergent lamivudine resistance-associated substitution (M184V or M184I), isolates from 0 of 22 subjects contained treatment-emergent amino acid substitutions associated with zidovudine resistance (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E), and isolates from 10 of 22 subjects contained treatment-emergent amino acid substitutions associated with efavirenz resistance (L100I, K101E, K103N, V108I, or Y181C).
Genotypic analysis of on-therapy isolates from subjects (n = 22) in the lamivudine twice-daily treatment group showed that isolates from 5 of 22 subjects contained treatment-emergent lamivudine resistance substitutions, isolates from 1 of 22 subjects contained treatment-emergent zidovudine resistance substitutions, and isolates from 7 of 22 subjects contained treatment-emergent efavirenz resistance substitutions.
Phenotypic analysis of baseline-matched on-therapy HIV-1 isolates from subjects (n = 13) receiving lamivudine once daily showed that isolates from 7 of 13 subjects showed an 85-to 299-fold decrease in susceptibility to lamivudine, isolates from 12 of 13 subjects were susceptible to zidovudine, and isolates from 8 of 13 subjects exhibited a 25-to 295-fold decrease in susceptibility to efavirenz.
Phenotypic analysis of baseline-matched on-therapy HIV-1 isolates from subjects (n = 13) receiving lamivudine twice daily showed that isolates from 4 of 13 subjects exhibited a 29-to 159-fold decrease in susceptibility to lamivudine, isolates from all 13 subjects were susceptible to zidovudine, and isolates from 3 of 13 subjects exhibited a 21-to 342-fold decrease in susceptibility to efavirenz.
Trial EPV40001: Fifty subjects received lamivudine 300 mg once daily plus zidovudine 300 mg twice daily plus abacavir 300 mg twice daily and 50 subjects received lamivudine 150 mg plus zidovudine 300 mg plus abacavir 300 mg all twice-daily. The median baseline plasma HIV-1 RNA levels for subjects in the 2 groups were 4.79 log10 copies per mL and 4.83 log10 copies per mL, respectively. Fourteen of 50 subjects in the lamivudine once-daily treatment group and 9 of 50 subjects in the lamivudine twice-daily group were identified as virologic failures.
Genotypic analysis of on-therapy HIV-1 isolates from subjects (n = 9) in the lamivudine once-daily treatment group showed that isolates from 6 subjects had an abacavir and/or lamivudine resistance-associated substitution M184V alone. On-therapy isolates from subjects (n = 6) receiving lamivudine twice daily showed that isolates from 2 subjects had M184V alone, and isolates from 2 subjects harbored the M184V substitution in combination with zidovudine resistance-associated amino acid substitutions.
Phenotypic analysis of on-therapy isolates from subjects (n = 6) receiving lamivudine once daily showed that HIV-1 isolates from 4 subjects exhibited a 32-to 53-fold decrease in susceptibility to lamivudine. HIV-1 isolates from these 6 subjects were susceptible to zidovudine.
Phenotypic analysis of on-therapy isolates from subjects (n = 4) receiving lamivudine twice daily showed that HIV-1 isolates from 1 subject exhibited a 45-fold decrease in susceptibility to lamivudine and a 4.5-fold decrease in susceptibility to zidovudine.
Tenofovir Disoproxil Fumarate: HIV-1 isolates with reduced susceptibility to tenofovir have been selected in cell culture. These viruses expressed a K65R substitution in RT and showed a 2- to 4-fold reduction in susceptibility to tenofovir. In addition, a K70E substitution in HIV-1 RT has been selected by tenofovir and results in low-level reduced susceptibility to tenofovir.
In Trial 903 of treatment-naïve subjects (tenofovir disoproxil fumarate + 3TC+EFV versus d4T+3TC+EFV, genotypic analyses of isolates from subjects with virologic failure through Week 144 showed development of EFV and 3TC resistance-associated substitutions to occur most frequently and with no difference between the treatment arms. The K65R substitution occurred in 8/47 (17%) of analyzed patient isolates in the tenofovir disoproxil fumarate arm and in 2/49 (4%) of analyzed patient isolates in the d4T arm. Of the 8 subjects whose virus developed K65R in the tenofovir disoproxil fumarate arm through 144 weeks, 7 occurred in the first 48 weeks of treatment and one at Week 96. One patient in the tenofovir disoproxil fumarate arm developed the K70E substitution in the virus. Other substitutions resulting in resistance to tenofovir disoproxil fumarate were not identified in this trial.
Cross-Resistance: Efavirenz: Cross-resistance among NNRTIs has been observed. Clinical isolates previously characterized as efavirenz-resistant were also phenotypically resistant in cell culture to delavirdine and nevirapine compared to baseline. Delavirdine- and/or nevirapine-resistant clinical viral isolates with NNRTI resistance-associated substitutions (A98G, L100I, K101E/P, K103N/S, V106A, Y181X, Y188X, G190X, P225H, F227L, or M230L) showed reduced susceptibility to efavirenz in cell culture. Greater than 90% of NRTI-resistant clinical isolates tested in cell culture retained susceptibility to efavirenz.
Lamivudine: Cross-resistance has been observed among nucleoside reverse transcriptase inhibitors (NRTIs). Lamivudine-resistant HIV-1 mutants were cross-resistant in cell culture to didanosine (ddI). Cross-resistance is also expected with abacavir and emtricitabine as these select M184V substitutions.
Tenofovir Disoproxil Fumarate: Cross resistance among certain HIV-1 NRTIs has been recognized. The K65R and K70E substitutions selected by tenofovir are also selected in some HIV-1 infected subjects treated with abacavir or didanosine. HIV-1 isolates with this substitution also show reduced susceptibility to FTC and 3TC. Therefore, cross resistance among these drugs may occur in patients whose virus harbors the K65R or K70E substitution. HIV-1 isolates from subjects (N=20) whose HIV-1 expressed a mean of three AZT-associated RT substitutions (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E/N) showed a 3.1-fold decrease in the susceptibility to tenofovir.
In Trials 902 and 907 conducted in treatment-experienced subjects (tenofovir disoproxil fumarate + Standard Background Therapy (SBT) compared to placebo + SBT) (see Pharmacokinetics as follows), 14/304 (5%) of the tenofovir disoproxil fumarate -treated subjects with virologic failure through Week 96 had >1.4-fold (median 2.7-fold) reduced susceptibility to tenofovir. Genotypic analysis of the baseline and failure isolates showed the development of the K65R substitution in the HIV-1 RT gene.
The virologic response to tenofovir disoproxil fumarate therapy has been evaluated with respect to baseline viral genotype (N=222) in treatment-experienced subjects participating in Trials 902 and 907. In these clinical trials, 94% of the participants evaluated had baseline HIV-1 isolates expressing at least one NRTI substitution. Virologic responses for subjects in the genotype sub study were similar to the overall trial results.
Several exploratory analyses were conducted to evaluate the effect of specific substitutions and substitutional patterns on virologic outcome. Because of the large number of potential comparisons, statistical testing was not conducted. Varying degrees of cross resistance of tenofovir disoproxil fumarate to pre-existing AZT resistance-associated substitutions (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E/N) were observed and appeared to depend on the type and number of specific substitutions. tenofovir disoproxil fumarate -treated subjects whose HIV-1 expressed 3 or more AZT resistance-associated substitutions that included either the M41L or L210W RT substitution showed reduced responses to tenofovir disoproxil fumarate therapy; however, these responses were still improved compared with placebo. The presence of the D67N, K70R, T215Y/F, or K219Q/E/N substitution did not appear to affect responses to tenofovir disoproxil fumarate therapy. Subjects whose virus expressed an L74V substitution without AZT resistance-associated substitutions (N=8) had reduced response to tenofovir disoproxil fumarate. Limited data are available for subjects whose virus expressed a Y115F substitution (N=3), Q151M substitution (N=2), or T69 insertion (N=4), all of whom had a reduced response.
Trials 902 and 907 Phenotypic Analyses: Phenotypic analysis of baseline HIV-1 from treatment-experienced subjects (N=100) demonstrated a correlation between baseline susceptibility to tenofovir disoproxil fumarate and response to tenofovir disoproxil fumarate therapy. Table 1 summarizes the HIV-1 RNA response by baseline tenofovir disoproxil fumarate susceptibility. (See Table 1.)
 Click on icon to see table/diagram/image
Pediatric Use:
Click on icon to see table/diagram/image
Pediatric Use: Efavirenz, lamivudine and tenofovir disoproxil fumarate tablets should only be administered to patients greater than 16 years of age with a body weight greater than or equal to 40 kg (greater than or equal to 88 lbs).
Geriatric Use: Clinical studies of efavirenz, lamivudine and tenofovir disoproxil fumarate did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other therapy.
Patients with Impaired Renal Function: Efavirenz, lamivudine and tenofovir disoproxil fumarate tablets are not recommended for patients with impaired renal function (i.e., creatinine clearance less than 50 mL/min) or patients with end-stage renal disease (ESRD) requiring hemodialysis because they are part of a fixed-dose combination formulation that cannot be adjusted.
Hepatic Impairment: Efavirenz, a component of efavirenz, lamivudine and tenofovir disoproxil fumarate, is not recommended for patients with moderate or severe hepatic impairment because there are insufficient data to determine whether dose adjustment is necessary. Patients with mild hepatic impairment may be treated with efavirenz without any adjustment in dose. Because of the extensive cytochrome P450-mediated metabolism of efavirenz and limited clinical experience in patients with hepatic impairment, caution should be exercised in administering efavirenz to these patients (see Precautions and Pharmacokinetics as follows).
Clinical Studies: Clinical Efficacy in Patients with HIV-1 Infection: Treatment-Naïve Subjects: Trial 903: Data through 144 weeks are reported for Trial 903, a double-blind, active-controlled multicenter trial comparing tenofovir disoproxil fumarate (300 mg once daily) administered in combination with lamivudine (3TC) and efavirenz (EFV) versus stavudine (d4T), 3TC, and EFV in 600 antiretroviral-naïve subjects. Subjects had a mean age of 36 years (range 18-64); 74% were male, 64% were Caucasian, and 20% were Black. The mean baseline CD4
+ cell count was 279 cells/mm
3 (range 3-956) and median baseline plasma HIV-1 RNA was 77,600 copies/mL (range 417-5,130,000). Subjects were stratified by baseline HIV-1 RNA and CD4
+ cell count. Forty-three percent of subjects had baseline viral loads >100,000 copies/mL and 39% had CD4
+ cell counts <200 cells/mm
3. Table 2 provides treatment outcomes through 48 and 144 weeks. (See Table 2.)
Click on icon to see table/diagram/image
Subjects achieved and maintained confirmed HIV-1 RNA <400 copies/mL through Week 48 and 144.
Includes confirmed viral rebound and failure to achieve confirmed <400 copies/mL through Week 48 and 144.
Includes lost to follow-up, subject's withdrawal, noncompliance, protocol violation and other reasons.
Achievement of plasma HIV-1 RNA concentrations of <400 copies/mL at Week 144 was similar between the two treatment groups for the population stratified at baseline on the basis of HIV-1 RNA concentration (> or ≤100,000 copies/mL) and CD4
+ cell count (< or ≥200 cells/mm
3). Through 144 weeks of therapy, 62% and 58% of subjects in the tenofovir disoproxil fumarate and d4T arms, respectively achieved and maintained confirmed HIV-1 RNA <50 copies/mL. The mean increase from baseline in CD4
+ cell count was 263 cells/mm
3 for the tenofovir disoproxil fumarate arm and 283 cells/mm
3 for the d4T arm.
Through 144 weeks, 11 subjects in the tenofovir disoproxil fumarate group and 9 subjects in the d4T group experienced a new CDC Class C event.
Pharmacokinetics: Efavirenz, lamivudine and tenofovir disoproxil fumarate tablets (600 mg, 300 mg, 300 mg) were bioequivalent to Sustiva (efavirenz, 600 mg tablet), Epivir (lamivudine 300 mg tablet) plus Viread (tenofovir disoproxil fumarate tablet) when administered to healthy volunteers under fasting conditions.
Efavirenz, lamivudine and tenofovir disoproxil fumarate tablets (600 mg, 300 mg, 300 mg) have not been evaluated under fed conditions. Efavirenz and products containing efavirenz should be administered under fasted conditions.
Lamivudine: The steady-state pharmacokinetic properties of the lamivudine 300-mg tablet once daily for 7 days compared with the lamivudine 150-mg tablet twice daily for 7 days were assessed in a crossover trial in 60 healthy subjects. EPIVIR 300 mg once daily resulted in lamivudine exposures that were similar to lamivudine 150 mg twice daily with respect to plasma AUC24,ss; however, Cmax,ss was 66% higher and the trough value was 53% lower compared with the 150-mg twice-daily regimen. Intracellular lamivudine triphosphate exposures in peripheral blood mononuclear cells were also similar with respect to AUC24,ss and Cmax24,ss; however, trough values were lower compared with the 150-mg twice-daily regimen. Inter-subject variability was greater for intracellular lamivudine triphosphate concentrations versus lamivudine plasma trough concentrations.
The pharmacokinetics of lamivudine was evaluated in 12 adult HIV-1-infected subjects dosed with lamivudine 150 mg twice daily in combination with other antiretroviral agents. The geometric mean (95% CI) for AUC(0-12) was 5.53 (4.58, 6.67) mcg.h per mL and for Cmax was 1.40 (1.17, 1.69) mcg per mL.
Absorption: Efavirenz: Peak efavirenz plasma concentrations of 1.6-9.1 jtM were attained by 5 hours following single oral doses of 100 mg to 1600 mg administered to uninfected volunteers. Dose-related increases in Cmax and AUC were seen for doses up to 1600 mg; the increases were less than proportional suggesting diminished absorption at higher doses.
In HIV-1-infected patients at steady state, mean Cmax, mean Cmin, and mean AUC were dose proportional following 200 mg, 400 mg, and 600 mg daily doses. Time-to-peak plasma concentrations were approximately 3-5 hours and steady-state plasma concentrations were reached in 6-10 days. In 35 patients receiving efavirenz 600 mg once daily, steady-state Cmax was 12.9 ± 3.7 jtM (mean ± SD), steady-state Cmin was 5.6 ± 3.2 jtM, and AUC was 184 ± 73 jtM·h.
Lamivudine: Absorption and Bioavailability: Absolute bioavailability in 12 adult subjects was 86% ± 16% (mean ± SD) for the 150-mg tablet and 87% ± 13% for the oral solution. After oral administration of 2 mg per kg twice a day to 9 adults with HIV-1, the peak serum lamivudine concentration (Cmax) was 1.5 ± 0.5 mcg per mL (mean ± SD). The area under the plasma concentration versus time curve (AUC) and Cmax increased in proportion to oral dose over the range from 0.25 to 10 mg per kg.
The accumulation ratio of lamivudine in HIV-1-positive asymptomatic adults with normal renal function was 1.50 following 15 days of oral administration of 2 mg per kg twice daily.
Tenofovir Disoproxil Fumarate: VIREAD is a water soluble diester prodrug of the active ingredient tenofovir. The oral bioavailability of tenofovir from VIREAD in fasted subjects is approximately 25%. Following oral administration of a single dose of VIREAD 300 mg to HIV-1 infected subjects in the fasted state, maximum serum concentrations (Cmax) are achieved in 1.0 ± 0.4 hrs. Cmax and AUC values are 0.30 ± 0.09 jtg/mL and 2.29 ± 0.69 jtg·hr/mL, respectively.
The pharmacokinetics of tenofovir are dose proportional over a VIREAD dose range of 75 to 600 mg and are not affected by repeated dosing.
In a single-dose bioequivalence study conducted under non-fasted conditions (dose administered with 4 oz. applesauce) in healthy adult volunteers, the mean Cmax of tenofovir was 26% lower for the oral powder relative to the tablet formulation. Mean AUC of tenofovir was similar between the oral powder and tablet formulations.
Distribution: Efavirenz: Efavirenz is highly bound (approximately 99.5-99.75%) to human plasma proteins, predominantly albumin. In HIV-1 infected patients (n=9) who received efavirenz 200 to 600 mg once daily for at least one month, cerebrospinal fluid concentrations ranged from 0.26 to 1.19% (mean 0.69%) of the corresponding plasma concentration. This proportion is approximately 3-fold higher than the non-protein-bound (free) fraction of efavirenz in plasma.
Lamivudine: Binding of lamivudine to human plasma proteins is less than 36%. In vitro studies showed that over the concentration range of 0.1 to 100 mcg per mL, the amount of lamivudine associated with erythrocytes ranged from 53% to 57% and was independent of concentration.
Tenofovir Disoproxil Fumarate: In vitro binding of tenofovir to human plasma or serum proteins is less than 0.7 and 7.2%, respectively, over the tenofovir concentration range 0.01 to 25 μg/mL.
Metabolism: Efavirenz: Studies in humans and in vitro studies using human liver microsomes have demonstrated that efavirenz is principally metabolized by the cytochrome P450 system to hydroxylated metabolites with subsequent glucuronidation of these hydroxylated metabolites. These metabolites are essentially inactive against HIV-1. The in vitro studies suggest that CYP3A and CYP2B6 are the major isozymes responsible for efavirenz metabolism.
Efavirenz has been shown to induce CYP enzymes, resulting in the induction of its own metabolism. Multiple doses of 200-400 mg per day for 10 days resulted in a lower than predicted extent of accumulation (22-42% lower) and a shorter terminal half-life of 40-55 hours (single dose half-life 52-76 hours).
Lamivudine: Metabolism of lamivudine is a minor route of elimination. In humans, the only known metabolite of lamivudine is the trans-sulfoxide metabolite (approximately 5% of an oral dose after 12 hours). Serum concentrations of this metabolite have not been determined. Lamivudine is not significantly metabolized by cytochrome P450 enzymes.
Tenofovir Disoproxil Fumarate: In vitro studies indicate that neither tenofovir disoproxil nor tenofovir are substrates of CYP enzymes.
Following single dose, oral administration of VIREAD, the terminal elimination half-life of tenofovir is approximately 17 hours. After multiple oral doses of VIREAD 300 mg once daily (under fed conditions), 32 ± 10% of the administered dose is recovered in urine over 24 hours.
Elimination: Efavirenz: Efavirenz has a terminal half-life of 52-76 hours after single doses and 40-55 hours after multiple doses. A one-month mass balance/excretion study was conducted using 400 mg per day with a C-labeled dose administered on Day 8. Approximately 14-34% of the radiolabel was recovered in the urine and 16-61% was recovered in the feces. Nearly all of the urinary excretion of the radiolabeled drug was in the form of metabolites. Efavirenz accounted for the majority of the total radioactivity measured in feces.
Lamivudine: The majority of lamivudine is eliminated unchanged in urine by active organic cationic secretion. In 9 healthy subjects given a single 300-mg oral dose of lamivudine, renal clearance was 199.7 ± 56.9 mL per min (mean ± SD). In 20 HIV-1-infected subjects given a single IV dose, renal clearance was 280.4 ± 75.2 mL per min (mean ± SD), representing 71% ± 16% (mean SD) of total clearance of lamivudine.
In most single-dose trials in HIV-1-infected subjects, HBV-infected subjects, or healthy subjects with serum sampling for 24 hours after dosing, the observed mean elimination half-life (t1/2) ranged from 5 to 7 hours. In HIV-1-infected subjects, total clearance was 398.5 ± 69.1 mL per min (mean ± SD). Oral clearance and elimination half-life were independent of dose and body weight over an oral dosing range of 0.25 to 10 mg per kg.
Tenofovir Disoproxil Fumarate: Tenofovir is eliminated by a combination of glomerular filtration and active tubular secretion. There may be competition for elimination with other compounds that are also renally eliminated.
Special Populations: Race: Efavirenz and Lamivudine: There are no significant racial differences in efavirenz and lamivudine pharmacokinetics.
Tenofovir Disoproxil Fumarate: There were insufficient numbers from racial and ethnic groups other than Caucasian to adequately determine potential pharmacokinetic differences among these populations.
Gender: There are no significant gender differences in the pharmacokinetics of efavirenz, lamivudine, and tenofovir disoproxil fumarate.
Geriatric Patients: The pharmacokinetics of lamivudine and tenofovir disoproxil fumarate have not been studied in patients over 65 years of age.
Pediatrics: Efavirenz: This combination tablet should not be administered to patients less than or equal to 16 years of age or patients weighing less than 40 kg.
The pharmacokinetic parameters for efavirenz at steady state in pediatric patients were predicted by a population pharmacokinetic model and are summarized in Table 3 by weight ranges that correspond to the recommended doses. (See Table 3.)
Click on icon to see table/diagram/image
Gender and race: The pharmacokinetics of efavirenz in patients appear to be similar between men and women and among the racial groups studied.
Renal impairment: The pharmacokinetics of efavirenz have not been studied in patients with renal insufficiency; however, less than 1% of efavirenz is excreted unchanged in the urine, so the impact of renal impairment on efavirenz elimination should be minimal.
Hepatic impairment: A multiple-dose study showed no significant effect on efavirenz pharmacokinetics in patients with mild hepatic impairment (Child-Pugh Class A) compared with controls. There were insufficient data to determine whether moderate or severe hepatic impairment (Child-Pugh Class B or C) affects efavirenz pharmacokinetics.
Tenofovir disoproxil fumarate: 2 Years and Older: Steady-state pharmacokinetics of tenofovir were evaluated in 31 HIV-1 infected pediatric subjects 2 years to less than 18 years of age (Table 4). Tenofovir exposure achieved in these pediatric subjects receiving oral once daily doses of tenofovir disoproxil fumarate 300 mg (tablet) or 8 mg/kg of body weight (powder) up to a maximum dose of 300 mg was similar to exposures achieved in adults receiving once-daily doses of tenofovir disoproxil fumarate 300 mg.
Tenofovir disoproxil fumarate: 2 Years and Older: Steady-state pharmacokinetics of tenofovir were evaluated in 31 HIV-1 infected pediatric subjects 2 years to less than 18 years of age (Table 4). Tenofovir exposure achieved in these pediatric subjects receiving oral once daily doses of tenofovir disoproxil fumarate 300 mg (tablet) or 8 mg/kg of body weight (powder) up to a maximum dose of 300 mg was similar to exposures achieved in adults receiving once-daily doses of tenofovir disoproxil fumarate 300 mg. (See Table 4.)
Click on icon to see table/diagram/image
Patients with Impaired Renal Function: Efavirenz, lamivudine and tenofovir disoproxil fumarate is not recommended for patients with impaired renal function (i.e., creatinine clearance less than 50 mL/min) or patients with end-stage renal disease (ESRD) requiring hemodialysis because it is a fixed-dose combination formulation that cannot be adjusted.
Patients with Hepatic Impairment: Efavirenz: A multiple-dose study showed no significant effect on efavirenz pharmacokinetics in patients with mild hepatic impairment (Child-Pugh Class A) compared with controls. There were insufficient data to determine whether moderate or severe hepatic impairment (Child-Pugh Class B or C) affects efavirenz pharmacokinetics.
Lamivudine: The pharmacokinetic properties of lamivudine have been determined in adults with impaired hepatic function. Pharmacokinetic parameters were not altered by diminishing hepatic function. Safety and efficacy of lamivudine have not been established in the presence of decompensated liver disease.
Tenofovir disoproxil fumarate: The pharmacokinetics of tenofovir following a 300 mg single dose of tenofovir disoproxil fumarate have been studied in non-HIV infected subjects with moderate to severe hepatic impairment. There were no substantial alterations in tenofovir pharmacokinetics in subjects with hepatic impairment compared with unimpaired subjects. No change in tenofovir disoproxil fumarate dosing is required in patients with hepatic impairment.
Assessment of Drug Interaction (see Interactions): Efavirenz: Efavirenz has been shown in vivo to cause hepatic enzyme induction, thus increasing the biotransformation of some drugs metabolized by CYP3A and CYP2B6. In vitro studies have shown that efavirenz inhibited CYP isozymes 2C9 and 2C19 with Ki values (8.5-17 μM) in the range of observed efavirenz plasma concentrations. In in vitro studies, efavirenz did not inhibit CYP2E1 and inhibited CYP2D6 and CYP1A2 (Ki values 82-160 μM) only at concentrations well above those achieved clinically. Coadministration of efavirenz with drugs primarily metabolized by CYP2C9, CYP2C19, CYP3A, or CYP2B6 isozymes may result in altered plasma concentrations of the coadministered drug. Drugs which induce CYP3A and CYP2B6 activity would be expected to increase the clearance of efavirenz resulting in lowered plasma concentrations.
Drug interaction studies were performed with efavirenz and other drugs likely to be coadministered or drugs commonly used as probes for pharmacokinetic interaction. The effects of coadministration of efavirenz on the Cmax, AUC, and Cmin are summarized in Table 5 (effect of efavirenz on other drugs) and Table 6 (effect of other drugs on efavirenz). For information regarding clinical recommendations see Interactions. (See Tables 5 and 6.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Lamivudine: Effect of Lamivudine on the Pharmacokinetics of Other Agents: Based on in vitro study results, lamivudine at therapeutic drug exposures is not expected to affect the pharmacokinetics of drugs that are substrates of the following transporters: organic anion transporter polypeptide 1B1/3 (OATP1B1/3), breast cancer resistance protein (BCRP), P-glycoprotein (P-gp), multidrug and toxin extrusion protein 1 (MATE)1, MATE2-K, organic cation transporter 1 (OCT)1, OCT2, or OCT3.
Effect of Other Agents on the Pharmacokinetics of Lamivudine: Lamivudine is a substrate of MATE1, MATE2-K, and OCT2 in vitro. Trimethoprim (an inhibitor of these drug transporters) has been shown to increase lamivudine plasma concentrations. This interaction is not considered clinically significant as no dose adjustment of lamivudine is needed.
Lamivudine is a substrate of P-gp and BCRP; however, considering its absolute bioavailability (87%), it is unlikely that these transporters play a significant role in the absorption of lamivudine. Therefore, coadministration of drugs that are inhibitors of these efflux transporters is unlikely to affect the disposition and elimination of lamivudine.
Interferon Alfa: There was no significant pharmacokinetic interaction between lamivudine and interferon alfa in a trial of 19 healthy male subjects (see Precautions).
Ribavirin: In vitro data indicate ribavirin reduces phosphorylation of lamivudine, stavudine, and zidovudine. However, no pharmacokinetic (e.g., plasma concentrations or intracellular triphosphorylated active metabolite concentrations) or pharmacodynamic (e.g., loss of HIV-1/HCV virologic suppression) interaction was observed when ribavirin and lamivudine (n = 18), stavudine (n = 10), or zidovudine (n = 6) were coadministered as part of a multi-drug regimen to HIV-1/HCV co-infected subjects (see Precautions).
Trimethoprim/Sulfamethoxazole: Lamivudine and TMP/SMX were coadministered to 14 HIV-1-positive subjects in a single-center, open-label, randomized, crossover trial. Each subject received treatment with a single 300-mg dose of lamivudine and TMP 160 mg/SMX 800 mg once a day for 5 days with concomitant administration of lamivudine 300 mg with the fifth dose in a crossover design. Coadministration of TMP/SMX with lamivudine resulted in an increase of 43% ± 23% (mean ± SD) in lamivudine AUC∞, a decrease of 29% ± 13% in lamivudine oral clearance, and a decrease of 30% ± 36% in lamivudine renal clearance. The pharmacokinetic properties of TMP and SMX were not altered by coadministration with lamivudine. There is no information regarding the effect on lamivudine pharmacokinetics of higher doses of TMP/SMX such as those used in treat PCP.
Zidovudine: No clinically significant alterations in lamivudine or zidovudine pharmacokinetics were observed in 12 asymptomatic HIV-1-infected adult subjects given a single dose of zidovudine (200 mg) in combination with multiple doses of lamivudine (300 mg every 12 hours).
Tenofovir Disoproxil Fumarate: At concentrations, substantially higher (~300-fold) than those observed in vivo, tenofovir did not inhibit in vitro drug metabolism mediated by any of the following human CYP isoforms: CYP3A4, CYP2D6, CYP2C9, or CYP2E1. However, a small (6%) but statistically significant reduction in metabolism of CYP1A substrate was observed. Based on the results of in vitro experiments and the known elimination pathway of tenofovir, the potential for CYP-mediated interactions involving tenofovir with other medicinal products is low.
Tenofovir disoproxil fumarate has been evaluated in healthy volunteers in combination with other antiretroviral and potential concomitant drugs. Tables 7 and 8 summarize pharmacokinetic effects of coadministered drug on tenofovir pharmacokinetics and effects of tenofovir disoproxil fumarate on the pharmacokinetics of coadministered drug.
TDF is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) transporters. When TDF is coadministered with an inhibitor of these transporters, an increase in absorption may be observed.
No clinically significant drug interactions have been observed between tenofovir disoproxil fumarate and efavirenz, methadone, nelfinavir, oral contraceptives, ribavirin, or sofosbuvir. (See Table 7.)
Click on icon to see table/diagram/image
No effect on the pharmacokinetic parameters of the following coadministered drugs was observed with tenofovir disoproxil fumarate: abacavir, didanosine (buffered tablets), emtricitabine, entecavir, and lamivudine. (See Table 8.)
Click on icon to see table/diagram/image


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image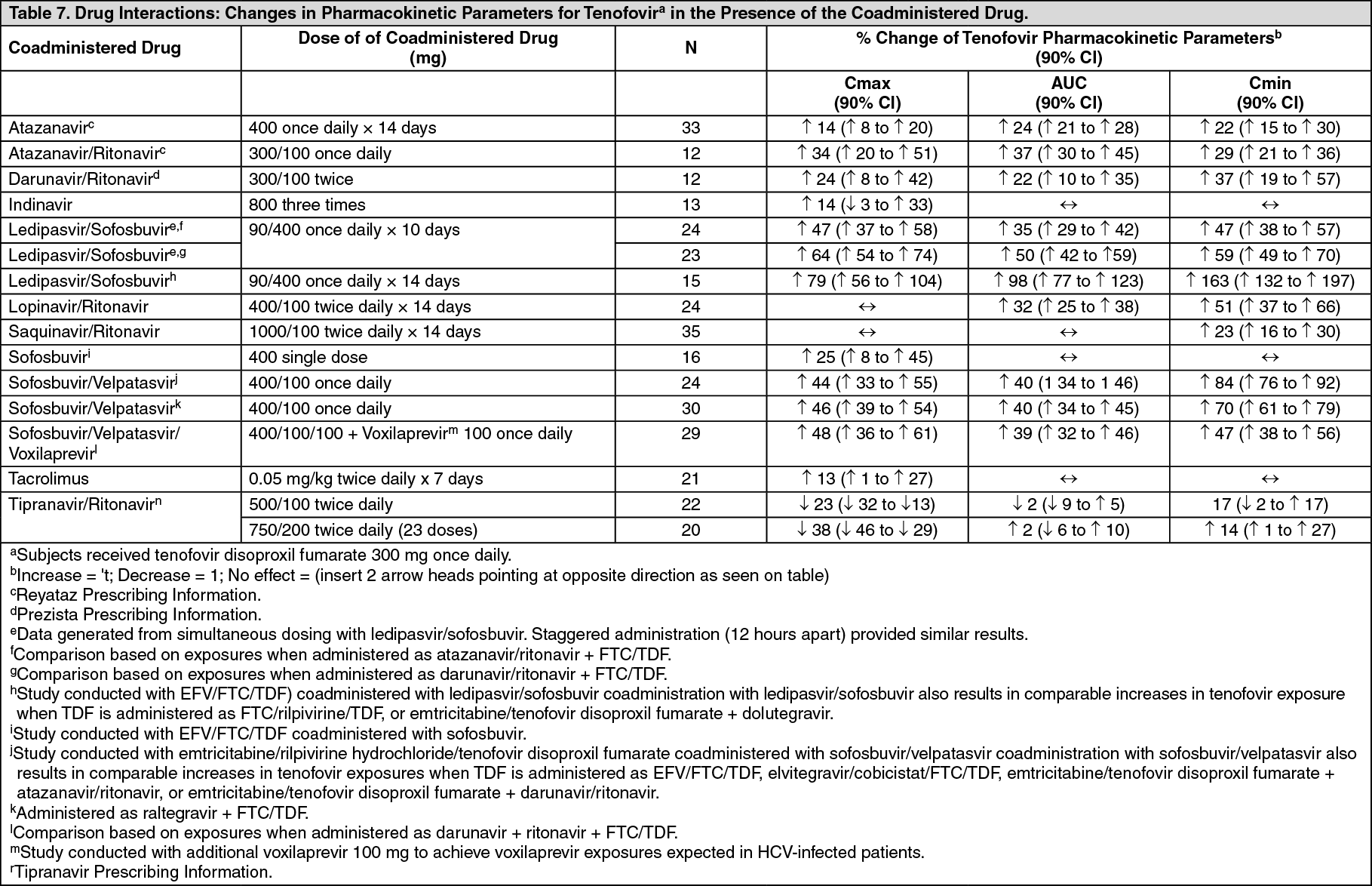 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image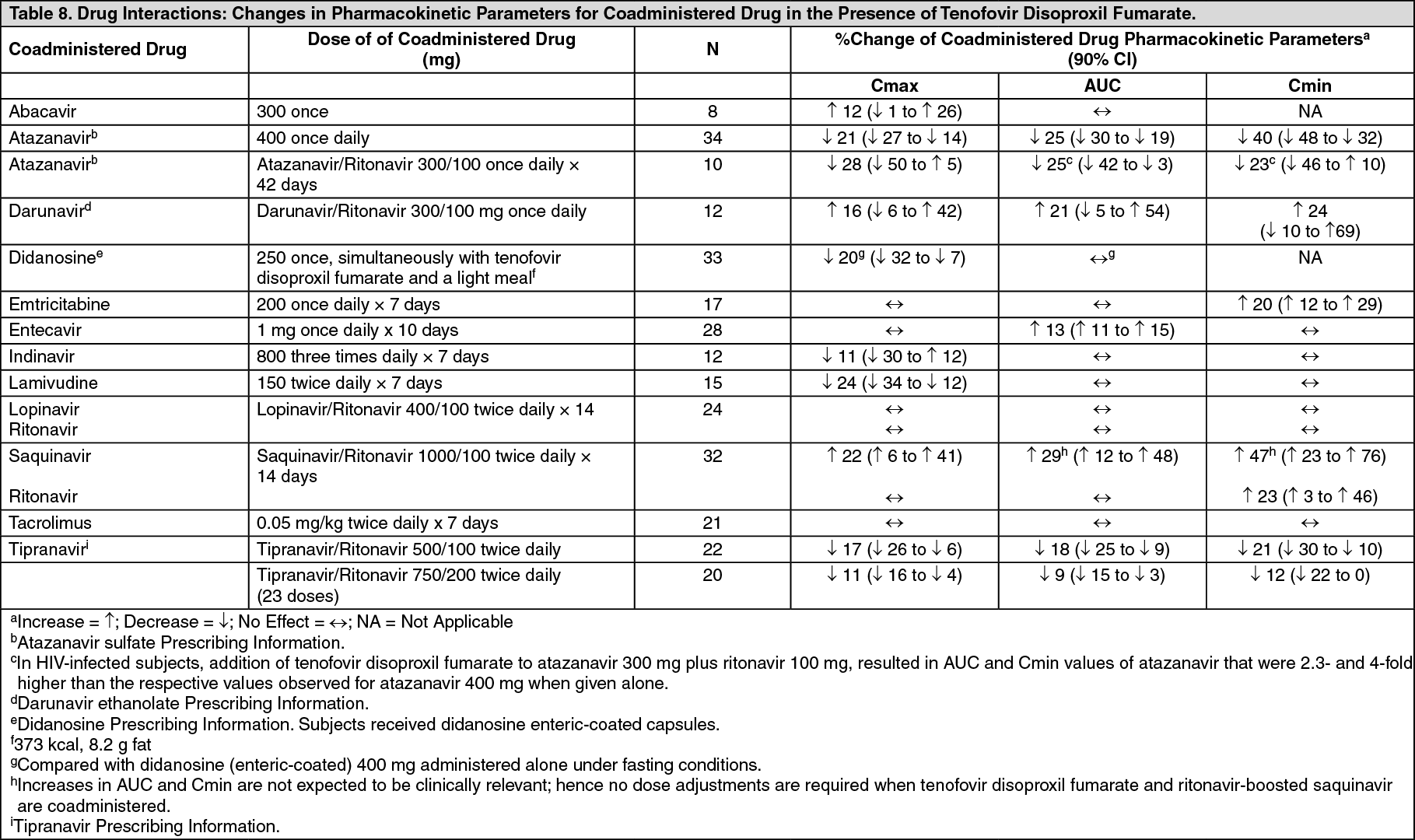 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
