Clinical Trials: Estradiol levels may increase during the first weeks following the initial injection of Leuprolide Acetate, but then decline to menopausal levels. This transient increase in estradiol can be associated with a temporary worsening of signs and symptoms (see Warnings).
As would be expected with a drug that lowers serum estradiol levels, the most frequently reported adverse reactions were those related to hypoestrogenism.
The monthly formulation of Leuprolide Acetate 3. 75 mg was utilized in controlled clinical trials that studied the drug in 166 endometriosis and 166 uterine fibroids patients. Adverse events reported in ≥5% of patients in either of these populations and thought to be potentially related to drug are noted in the following table. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In one controlled clinical trial utilizing the monthly formulation of leuprorelin acetate, patients diagnosed with uterine fibroids received a higher dose (7.5 mg) of leuprorelin acetate. Events seen with this dose that were thought to be potentially related to drug and were not seen at the lower dose included glossitis, hypesthesia, lactation, pyelonephritis, and urinary disorders. Generally, a higher incidence of hypoestrogenic effects was observed at the higher dose.
Table 3 lists the potentially drug-related adverse events observed in at least 5% of patients in any treatment group during the first 6 months of treatment in the add-back clinical studies.
In the controlled clinical trial, 50 of 51 (98%) patients in the LD group and 48 of 55 (87%) patients in the LD/N group reported experiencing hot flashes on one or more occasions during treatment. During Month 6 of treatment, 32 of 37 (86%) patients in the LD group and 22 of 38 (58%) patients in the LD/N group reported having experienced hot flashes. The mean number of days on which hot flashes were reported during this month of treatment was 19 and 7 in the LO and LOIN treatment groups, respectively. The mean maximum number of hot flashes in a day during this month of treatment was 5.8 and 1.9 in the LD and LD/N treatment groups, respectively. (See Table 3.)
Click on icon to see table/diagram/image
Changes in Bone Density: In controlled clinical studies, patients with endometriosis (six months of therapy) or uterine fibroids (three months of therapy) were treated with leuprorelin acetate 3.75 mg. In endometriosis patients, vertebral bone density as measured by dual energy x-ray absorptiometry (DEXA) decreased by an average of 3.2% at six months compared with the pretreatment value. Clinical studies demonstrate that concurrent hormonal therapy (norethindrone acetate 5 mg daily) and calcium supplementation is effective in significantly reducing the loss of bone mineral density that occurs with leuprorelin acetate treatment, without compromising the efficacy of leuprorelin acetate in relieving symptoms of endometriosis.
Leuprorelin acetate plus norethindrone acetate 5 mg daily was evaluated in two clinical trials. The results from this regimen were similar in both studies. Prolide was used as a control group in one study. The bone mineral density data of the lumbar spine from these two studies are presented in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
When leuprorelin acetate was administered for three months in uterine fibroid patients, vertebral trabecular bone mineral density as assessed by quantitative digital radiography (QDR) revealed a mean decrease of 2.7% compared with baseline. Six months after discontinuation of therapy, a trend toward recovery was observed. Use of Prolide for longer than three months (uterine fibroids) or six months (endometriosis) or in the presence of other known risk factors for decreased bone mineral content may cause additional bone loss and is not recommended.
Changes In Laboratory Values During Treatment: Plasma Enzymes: Endometriosis: During early clinical trials with leuprorelin acetate, regular laboratory monitoring revealed that AST levels were more than twice the upper limit of normal in only one patient. There was no clinical or other laboratory evidence of abnormal liver function.
In two other clinical trials, 6 of 191 patients receiving Leuprolide Acetate 3. 75 mg plus norethindrone acetate 5 mg daily for up to 12 months developed an elevated (at least twice the upper limit of normal) SGPT or GGT. Five of the 6 increases were observed beyond 6 months of treatment. None were associated with elevated bilirubin concentration.
Uterine Leiomyomata (Fibroids): In clinical trials with leuprorelin acetate, five (3%) patients had a post-treatment transaminase value that was at least twice the baseline value and above the upper limit of the nominal range. None of the laboratory increases were associated with clinical symptoms.
Lipids: Endometriosis: In earlier clinical studies, 4% of the leuprorelin acetate patients and 1% of the danazol patients had total cholesterol values above the normal range at enrollment. These patients also had cholesterol values above the normal range at the end of treatment.
Of those patients whose pretreatment cholesterol values were in the normal range, 7% of the leuprorelin acetate patients and 9% of the danazol patients had post-treatment values above the normal range.
The mean (±SEM) pretreatment values for total cholesterol from all patients were 178.8 (2.9) mg/dL in the leuprorelin acetate groups and 175.3 (3.0) mg/dL in the danazol group. At the end of treatment, the mean values for total cholesterol from all patients were 193.3 mg/dL in the leuprorelin acetate group and 194.4 mg/dL in the danazol group. These increases from the pretreatment values were statistically significant (p<0.03) in both groups.
Triglycerides were increased above the upper limit of normal in 12% of the patients who received leuprorelin acetate and in 6% of the patients who received danazol.
At the end of treatment, HDL cholesterol fractions decreased below the lower limit of the normal range in 2% of the leuprorelin acetate patients compared with 54% of those receiving danazol. LDL cholesterol fractions increased above the upper limit of the normal range in 6% of the patients receiving leuprorelin acetate compared with 23% of those receiving danazol. There was no increase in the LDL/HDL ratio in patients receiving leuprorelin acetate but there was approximately a two-fold increase in the LDL/HDL ratio in patients receiving danazol.
In two other clinical trials, leuprorelin acetate plus norethindrone acetate 5 mg daily was evaluated for 12 months of treatment. leuprorelin acetate was used as a control group in one study. Percent changes from baseline for serum lipids and percentages of patients with serum lipid values outside of the nominal range in the two studies are summarized in the tables as follows. (See Table 5.)
Click on icon to see table/diagram/image
Changes from baseline tended to be greater at Week 52. After treatment, mean serum lipid levels from patients with follow up data returned to pretreatment values. (See Table 6.)
Click on icon to see table/diagram/image
Low HDL-cholesterol (<40 mg/dL) and elevated LDL-cholesterol (>160 mg/dL) are recognized risk factors for cardiovascular disease. The long-term significance of the observed treatment-related changes in serum lipids in women with endometriosis is unknown. Therefore assessment of cardiovascular risk factors should be considered prior to initiation of concurrent treatment with leuprorelin acetate and norethindrone acetate.
Uterine Leiomyomata (Fibroids): In patients receiving Prolide , mean changes in cholesterol (+11 mg/dL to +29 mg/dL), LDL cholesterol (+8 mg/dL to +22 mg/dL), HDL cholesterol (0 to +6 mg/dL), and the LDL/HDL ratio (-0.1 to +0.5)were observed across studies. In the one study in which triglycerides were determined, the mean increase from baseline was 32 mg/dL.
Other Changes: Endometriosis: The following changes were seen in approximately 5% to 8% of patients. In the earlier comparative studies, leuprorelin acetate was associated with elevations of LOH and phosphorus, and decreases in WBC counts. Danazol therapy was associated with increases in hematocrit, platelet count, and LOH. In the hormonal add-back studies leuprorelin acetate in combination with norethindrone acetate was associated with elevations of GGT and SGPT.
Uterine Leiomyomata (Fibroids): Hematology: (see Clinical studies as previously mentioned section). In Leuprorelin Acetate treated patients, although there were statistically significant mean decreases in platelet counts from baseline to final visit, the last mean platelet counts were within the normal range. Decreases in total WBC count and neutrophils were observed, but were not clinically significant.
Chemistry: Slight to moderate mean increases were noted for glucose, uric acid, BUN, creatinine, total protein, albumin, bilirubin, alkaline phosphatase, LOH, calcium, and phosphorus. None of these increases were clinically significant.
Postmarketing: During postmarketing surveillance, the following adverse events were reported. Like other drugs in this class, mood swings, including depression, have been reported. There have been rare reports of suicidal ideation and attempt. Many, but not all, of these patients had a history of depression or other psychiatric illness. Patients should be counseled on the possibility of development or worsening of depression during treatment with Leuprorelin Acetate.
Symptoms consistent with an anaphylactoid or asthmatic process have been rarely reported. Rash, urticaria, and photosensitivity reactions have also been reported.
Localized reactions including induration and abscess have been reported at the site of injection. Symptoms consistent with fibromyalgia (eg: joint and muscle pain, headaches, sleep disorder, gastrointestinal distress, and shortness of breath) have been reported individually and collectively.
Other events reported are:
Cardiovascular System- Hypotension;
Cases of serious venous and arterial thromboembolism have been reported, including deep vein thrombosis, pulmonary embolism, myocardial infarction, stroke, and transient ischemic attack. Although a temporal relationship was reported in some cases, most cases were confounded by risk factors or concomitant medication use. It is unknown if there is a causal association between the use of GnRH analogs and these events.
Hemic And Lymphatic System- Decreased WBC;
Central/Peripheral Nervous System- Convulsion, Peripheral neuropathy, Spinal fracture/paralysis; Musculoskeletal System- Tenosynovitis-like symptoms;
Urogenital System - Prostate pain.
Pituitary apoplexy.
During post-marketing surveillance, rare cases of pituitary apoplexy (a clinical syndrome secondary to infarction of the pituitary gland) have been reported after the administration of gonadotropin-releasing hormone agonists. In a majority of these cases, a pituitary adenoma was diagnosed, with a majority of pituitary apoplexy cases occurring within 2 weeks of the first dose, and some within the first hour. In these cases, pituitary apoplexy has presented as sudden headache, vomiting, visual changes, ophthalmoplegia, altered mental status, and sometimes cardiovascular collapse. Immediate medical attention has been required.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image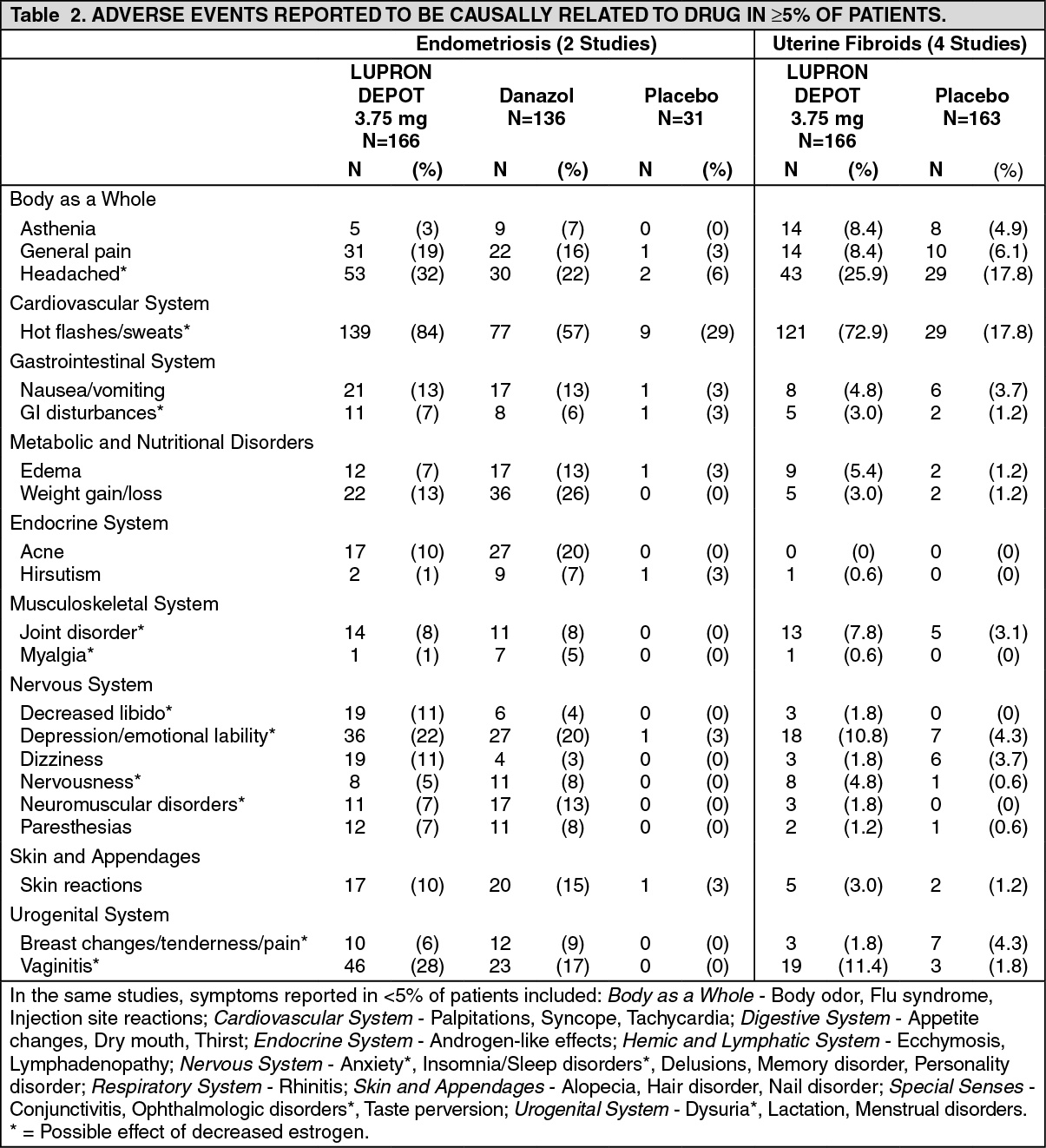 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image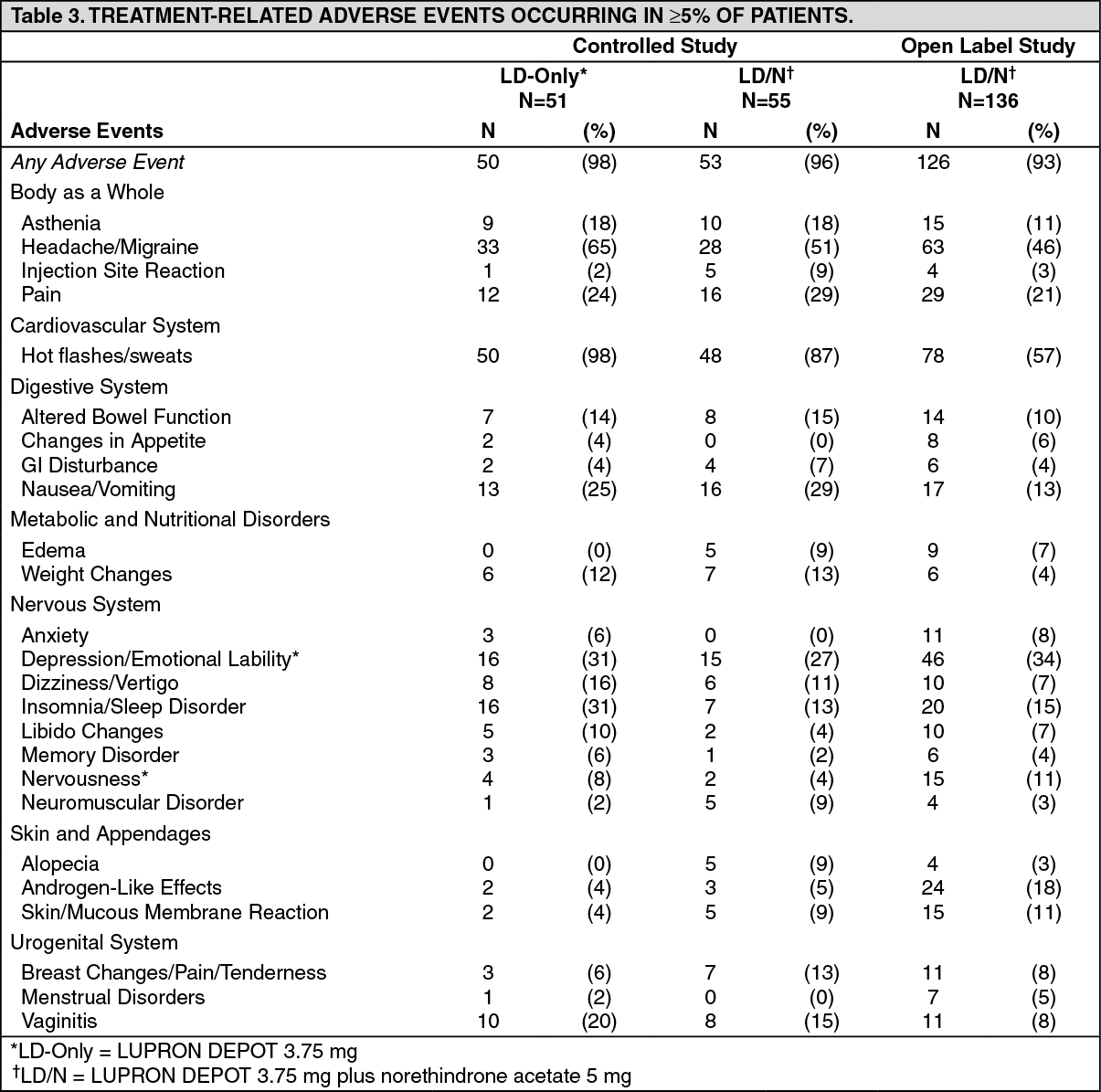 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image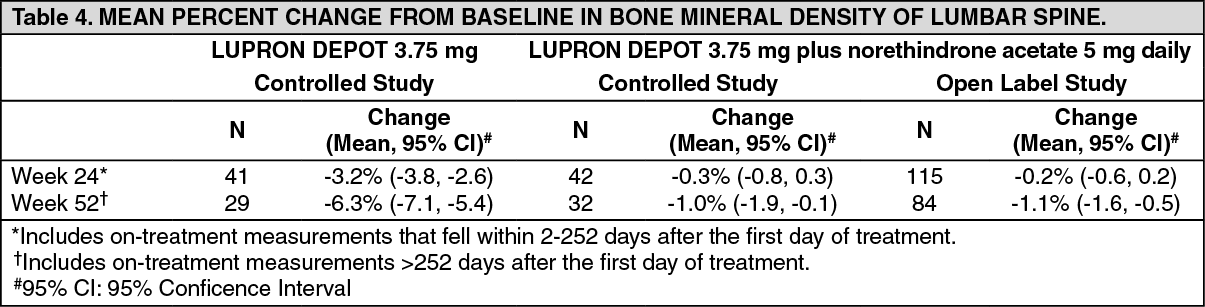 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image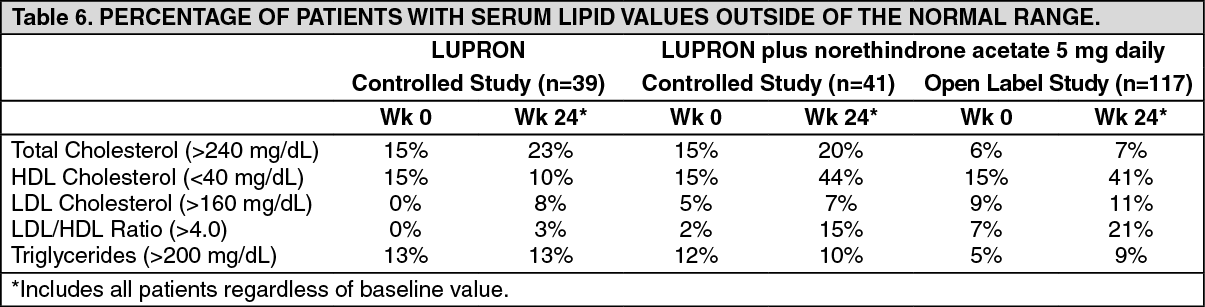 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
