


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of Action: Uric acid is the end product of purine metabolism in humans and is generated in the cascade of hypoxanthine → xanthine → uric acid. Both steps in the previously mentioned transformations are catalyzed by xanthine oxidase (XO). Febuxostat is a 2-arylthiazole derivative that achieves its therapeutic effect of decreasing serum uric acid by selectively inhibiting XO. Febuxostat is a potent, non-purine selective inhibitor of XO (NP-SIXO) with an in vitro inhibition Ki value less than 1 nanomolar. Febuxostat has been shown to potently inhibit both the oxidized and reduced forms of XO. At therapeutic concentrations Febuxostat does not inhibit other enzymes involved in purine or pyrimidine metabolism, namely, guanine deaminase, hypoxanthine guanine phosphoribosyltransferase, orotate phosphoribosyltransferase, orotidine monophosphate decarboxylase or purine nucleoside phosphorylase.
Clinical Efficacy: The efficacy of Febuxostat was demonstrated in three Phase 3 pivotal studies (the two pivotal APEX and FACT studies, and the additional CONFIRMS study described as follows) that were conducted in 4,101 patients with hyperuricemia and gout. In each phase 3 pivotal study, Febuxostat demonstrated superior ability to lower and maintain serum uric acid levels compared to allopurinol. The primary efficacy endpoint in the APEX and FACT studies was the proportion of patients whose last 3 monthly serum uric acid levels were <6.0 mg/dL. In the additional phase 3 CONFIRMS study, for which results became available after the marketing authorization for Febuxostat was first issued, the primary efficacy endpoint was the proportion of patients whose serum urate level was <6.0 mg/dL at the final visit.
APEX Study: The Allopurinol and Placebo-Controlled Efficacy Study of Febuxostat (APEX) was a Phase 3, randomized, double-blind, multicenter, 28-week study. One thousand and seventy-two (1,072) patients were randomized: placebo (n=134), Febuxostat 80 mg QD (n=267), Febuxostat 120 mg QD (n=269), Febuxostat 240 mg QD (n=134) or allopurinol (300 mg QD [n=258] for patients with a baseline serum creatinine ≤1.5 mg/dL or 100 mg QD [n=10] for patients with a baseline serum creatinine >1.5 mg/dL and ≤2.0 mg/dL). 240 mg Febuxostat (2 times the recommended highest dose) was used as a safety evaluation dose. The APEX study showed statistically significant superiority of both the Febuxostat 80 mg QD and the Febuxostat 120 mg QD treatment arms versus the conventionally used doses of allopurinol 300 mg (n = 258)/100 mg (n = 10) treatment arm in reducing the sUA below 6 mg/dL (see table and figure).
FACT Study: The Febuxostat Allopurinol Controlled Trial (FACT) Study was a Phase 3, randomized, double-blind, multicenter, 52-week study. Seven hundred sixty (760) patients were randomized: Febuxostat 80 mg QD (n=256), Febuxostat 120 mg QD (n=251), or allopurinol 300 mg QD (n=253). The FACT study showed the statistically significant superiority of both Febuxostat 80 mg and Febuxostat 120 mg QD treatment arms versus the conventionally used dose of allopurinol 300 mg treatment arm in reducing and maintaining sUA below 6 mg/dL.
The table summarizes the primary efficacy endpoint results: See table.
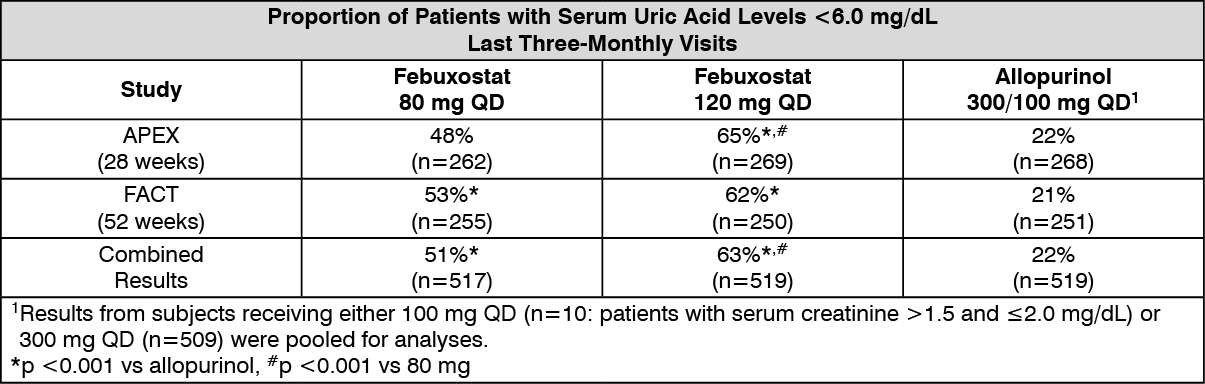 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe ability of Febuxostat to lower serum uric acid levels was prompt and persistent. Reduction in serum uric acid level to <6.0 mg/dL was noted by the Week 2 visit and was maintained throughout treatment. The mean serum uric acid levels over time for each treatment group from the two pivotal Phase 3 studies are shown in the figure.
Note: 509 patients received allopurinol 300 mg QD; 10 patients with serum creatinine >1.5 and <2.0 mg/dL were dosed with 100 mg QD. (10 patients out of 268 in APEX study).
240 mg Febuxostat was used to evaluate the safety of Febuxostat at twice the recommended highest dose.
CONFIRMS Study: The CONFIRMS study was a Phase 3, randomized, controlled, 26-week study to evaluate the safety and efficacy of Febuxostat 40 mg and 80 mg, in comparison with allopurinol 300 mg or 200 mg, in patients with gout and hyperuricemia. Two thousand and two hundred-sixty-nine (2,269) patients were randomized: Febuxostat 40 mg QD (n=757), Febuxostat 80 mg QD (n=756), or allopurinol 300/200 mg QD (n=756). At least 65% of the patients had mild-moderate renal impairment (with creatinine clearance of 30-89 mL/min). Prophylaxis against gout flares was obligatory over the 26-week period. The proportion of patients with serum urate levels of <6.0 mg/dL at the final visit, was 45% for 40 mg Febuxostat, 67% for Febuxostat 80 mg and 42% for allopurinol 300/200 mg, respectively.
Primary endpoint in the sub-group of patients with renal impairment: The APEX Study evaluated efficacy in 40 patients with renal impairment (i.e. baseline serum creatinine >1.5 mg/dL and ≤2.0 mg/dL). For renally impaired subjects who were randomized to allopurinol, the dose was capped at 100 mg QD. Febuxostat achieved the primary efficacy endpoint in 44% (80 mg QD), 45% (120 mg QD) and 60% (240 mg QD) of patients compared to 0% in the allopurinol 100 mg QD and placebo groups. There were no clinically significant differences in the percent decrease in serum uric acid concentration in healthy subjects irrespective of their renal function (58% in the normal renal function group and 55% in the severe renal dysfunction group). An analysis in patients with gout and renal impairment was prospectively defined in the CONFIRMS study, and showed that Febuxostat was significantly more efficacious in lowering serum urate levels to <6 mg/dL compared to allopurinol 300 mg/200 mg in patients who had gout with mild to moderate renal impairment (65% of patients studied).
Primary endpoint in the sub group of patients with sUA ≥10 mg/dL: Approximately 40% of patients (combined APEX and FACT) had a baseline sUA ≥10 mg/dL. In this subgroup, Febuxostat achieved the primary efficacy endpoint (sUA <6.0 mg/dL at the last 3 visits) in 41% (80 mg QD), 48% (120 mg QD), and 66% (240 mg QD) of patients compared to 9% in the allopurinol 300 mg/100 mg QD and 0% in the placebo groups.
In the CONFIRMS study, the proportion of patients achieving the primary efficacy endpoint (sUA <6.0 mg/dL at the final visit) for patients with a baseline serum urate level of ≥10 mg/dL treated with Febuxostat 40 mg QD was 27% (66/249), with Febuxostat 80 mg QD 49% (125/254) and with allopurinol 300 mg/200 mg QD 31% (72/230), respectively.
Clinical Outcomes: proportion of patients requiring treatment for a gout flare: APEX study: During the 8-week prophylaxis period, a greater proportion of subjects in the Febuxostat 120 mg (36%) treatment group required treatment for gout flare compared to Febuxostat 80 mg (28%), allopurinol 300 mg (23%) and placebo (20%). Flares increased following the prophylaxis period and gradually decreased overtime. Between 46% and 55% of subjects received treatment for gout flares from Week 8 and Week 28. Gout flares during the last 4 weeks of the study (Weeks 24-28) were observed in 15% (Febuxostat 80, 120 mg), 14% (allopurinol 300 mg) and 20% (placebo) of subjects.
FACT study: During the 8-week prophylaxis period, a greater proportion of subjects in the Febuxostat 120 mg (36%) treatment group required treatment for a gout flare compared to both the Febuxostat 80 mg (22%) and allopurinol 300 mg (21%) treatment groups. After the 8-week prophylaxis period, the incidences of flares increased and gradually decreased over time (64% and 70% of subjects received treatment for gout flares from Week 8-52). Gout flares during the last 4 weeks of the study (Weeks 49-52) were observed in 6-8% (Febuxostat 80 mg, 120 mg) and 11% (allopurinol 300 mg) of subjects. (See figure.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe proportion of subjects requiring treatment for a gout flare (APEX and FACT Study) was numerically lower in the groups that achieved an average post-baseline serum urate level <6.0 mg/dL, <5.0 mg/dL or <4.0 mg/dL compared to the group that achieved an average post-baseline serum urate level ≥6.0 mg/dL during the last 32 weeks of the treatment period (Week 20-Week 24 to Week 49-52 intervals). During the CONFIRMS study, the percentages of patients who required treatment for gout flares (Day 1 through Month 6) were 31% and 25% for the Febuxostat 80 mg and allopurinol groups, respectively. No difference in the proportion of patients requiring treatment for gout flares was observed between the Febuxostat 80 mg and 40 mg groups.
Long-term, open label extension Studies: EXCEL Study: The Excel study was a three-year Phase 3, open label, multicenter, randomized, allopurinol-controlled, safety extension study for patients who had completed the pivotal Phase 3 studies (APEX or FACT). A total of 1,086 patients were enrolled: Febuxostat 80 mg QD (n=649), Febuxostat 120 mg QD (n=292) and allopurinol 300/100 mg QD (n=145). About 69% of patients required no treatment change to achieve a final stable treatment. Patients who had 3 consecutive sUA levels >6.0 mg/dL were withdrawn. Serum urate levels were maintained overtime (i.e. 91% and 93% of patients on initial treatment with Febuxostat 80 mg and 120 mg, respectively, had sUA <6 mg/dL at Month 36). Three years data showed a decrease in the incidence of gout flares with less than 4% of patients requiring treatment for a flare (i.e. more than 96% of patients did not require treatment for a flare) at Month 16-24 and at Month 30-36. 46% and 38%, of patients on final stable treatment of Febuxostat 80 or 120 mg QD, respectively, had complete resolution of the primary palpable tophus from baseline to the Final Visit.
FOCUS Study was a 5-year Phase 2, open-label, multicenter, safety extension study for patients who had completed the Febuxostat 4 weeks of double-blind dosing in study TMX-00-004.
116 patients were enrolled and received initially Febuxostat 80 mg QD. 62% of patients required no dose adjustment to maintain sUA <6 mg/dL and 38% of patients required a dose adjustment to achieve a final stable dose. The proportion of patients with serum urate levels of <6.0 mg/dL at the final visit was greater than 80% (81-100%) at each Febuxostat dose.
During the phase 3 clinical studies, mild liver function test abnormalities were observed in patients treated with Febuxostat (5.0%). These rates were similar to the rates reported on allopurinol (4.2%). Increased TSH values(>5.5 μIU/mL) were observed in patients on long-term treatment with Febuxostat (5.5%) and patients with allopurinol (5.8%)in the long-term open label extension studies.
Post Marketing long term studies: CARES Study was a multi-center, randomized, double-blind, non-inferiority trial comparing CV outcomes with Febuxostat versus allopurinol in patients with gout and a history of major CV disease including MI, hospitalization for unstable angina, coronary or cerebral revascularization procedure, stroke, hospitalized transient ischemic attack, peripheral vascular disease, or diabetes mellitus with evidence of microvascular or macrovascular disease. To achieve sUA less than 6mg/dL, the dose of Febuxostat was titrated from 40mg up to 80mg (regardless of renal function) and the dose of allopurinol was titrated in 100mg increments from 300 to 600mg in patients with normal renal function and mild renal impairment and from 200 to 400mg in patients with moderate renal impairment.
The primary endpoint in CARES was the time to first occurrence of MACE, a composite of non-fatal MI, non-fatal stroke, CV death and unstable angina with urgent coronary revascularization.
The endpoints (primary and secondary) were analyzed according to the intention-to-treat (ITT) analysis including all subjects who were randomized and received at least one dose of double-blind study medication. Overall, 56.6% of patients discontinued trial treatment prematurely and 45% of patients did not complete all trial visits. In total, 6,190 patients were followed for a median of 32 months and the median duration of exposure was 728 days for patients in Febuxostat group (n=3098) and 719 days in allopurinol group (n=3092). The primary MACE endpoint occurred at similar rates in the Febuxostat and allopurinol treatment groups (10.8% vs. 10.4% of patients, respectively; hazard ratio [HR] 1.03; two-sided repeated 95% confidence interval [Cl] 0.87-1.23).
In the analysis of the individual components of MACE, the rate of CV deaths was higher with Febuxostat than allopurinol (4.3% vs. 3.2% of patients; HR 1.34; 95% Cl 1.03-1.73). The rates of the other MACE events were similar in the Febuxostat and allopurinol groups, i.e. non-fatal MI (3.6% vs. 3.8% of patients; HR 0.93; 95% Cl 0.72-1.21), non-fatal stroke (2.3% vs. 2.3% of patients; HR 1.01; 95% Cl 0.73-1.41) and urgent revascularization due to unstable angina (1.6% vs. 1.8% of patients; HR 0.86; 95% Cl 0.59-1.26). The rate of all-cause mortality was also higher with Febuxostat than allopurinol (7.8% vs. 6.4% of patients; HR 1.22; 95% Cl 1.01-1.47), which was mainly driven by the higher rate of CV deaths in that group. Rates of adjudicated hospitalization for heart failure, hospital admissions for arrhythmias not associated with ischemia, venous thromboembolic events and hospitalization for transient ischemic attacks were comparable for Febuxostat and allopurinol.
Pharmacokinetics: In healthy subjects, maximum plasma concentrations (Cmax) and area under the plasma concentration-time curve (AUC) of Febuxostat increased in a dose proportional manner following single and multiple doses of 10 mg to 120 mg. There is no appreciable accumulation when doses of 10 mg to 240 mg are administered every 24 hours. Febuxostat has an apparent mean terminal elimination half-life (t½) of approximately 5 to 8 hours. In general, Febuxostat pharmacokinetic parameters are consistent with those obtained from healthy subjects, indicating that healthy subjects are representative for pharmacokinetic/pharmacodynamic assessment in the patient population with gout.
Absorption: Febuxostat is rapidly (tmax of 1.0-1.5 h) and well absorbed (at least 84%). After single or multiple oral 80 and 120 mg once daily doses, Cmax is approximately 2.8-3.2 μg/mL, and 5.0-5.3 μg/mL, respectively. Absolute bioavailability of the Febuxostat tablet formulation has not been studied.
Following multiple oral 80 mg once daily doses or a single 120 mg dose with a high fat meal, there was a 49% and 38% decrease in Cmax and a 18% and 16% decrease in AUC, respectively. However, no clinically significant change in the percent decrease in serum uric acid concentration was observed where tested (80 mg multiple dose). Thus, Febuxostat Tablets may be taken without regard to food.
Distribution: The apparent steady-state volume of distribution (Vss/F) of Febuxostat ranges from 29 to 75 L after oral doses of 10-300 mg. The plasma protein binding of Febuxostat is approximately 99.2%, (primarily to albumin), and is constant over the concentration range achieved with 80 and 120 mg doses. Plasma protein binding of the active metabolites ranges from about 82% to 91%.
Biotransformation: Febuxostat is extensively metabolized by conjugation via uridine diphosphate glucuronosyltransferase (UDPGT) enzyme system and oxidation via the cytochrome P450 (CYP) system. Four pharmacologically active hydroxyl metabolites have been identified, of which three occur in plasma of humans. In vitro studies with human liver microsomes showed that those oxidative metabolites were formed primarily by CYP1A1, CYP1A2, CYP2C8 or CYP2C9 and Febuxostat glucuronide was formed mainly by UGT 1A1, 1A8, and 1A9.
Elimination: Febuxostat is eliminated by both hepatic and renal pathways. Following an 80 mg oral dose of 14C-labeled Febuxostat, approximately 49% of the dose was recovered in the urine as unchanged Febuxostat (3%), the acyl glucuronide of the active substance (30%), its known oxidative metabolites and their conjugates (13%), and other unknown metabolites (3%). In addition to the urinary excretion, approximately 45% of the dose was recovered in the feces as the unchanged Febuxostat (12%), the acyl glucuronide of the active substance (1%), its known oxidative metabolites and their conjugates (25%), and other unknown metabolites (7%).
Renal impairment: Following multiple doses of 80 mg of Febuxostat in patients with mild, moderate or severe renal impairment, the Cmax of Febuxostat did not change, relative to subjects with normal renal function. The mean total AUC of Febuxostat increased by approximately 1.8-foldfrom 7.5 μg·h/mL in the normal renal function group to 13.2 μg·h/mL in the severe renal dysfunction group. The Cmax and AUC of active metabolites increased up to 2- and 4-fold, respectively. However, no dose adjustment is necessary in patients with mild or moderate renal impairment.
Hepatic impairment: Following multiple doses of 80 mg of Febuxostat in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment, the Cmax and AUC of Febuxostat and its metabolites did not change significantly compared to subjects with normal hepatic function. No studies have been conducted in patients with severe hepatic impairment (Child-Pugh Class C).
Age: There were no significant changes observed in AUC of Febuxostat or its metabolites following multiple oral doses of Febuxostat in elderly was compared to younger healthy subjects.
Gender: Following multiple oral doses of Febuxostat, the Cmax and AUC were 24% and 12% higher in females than in males, respectively. However, weight-corrected Cmax and AUC were similar between the genders. No dose adjustment is needed based on gender.
Non-Clinical Toxicology: Effects in non-clinical studies were generally observed at exposures in excess of the maximum human exposure. Pharmacokinetic modelling and simulation of rat data suggests that, when co-administered with Febuxostat, the clinical dose of mercaptopurine/azathioprine should be reduced to 20% or less of the previously prescribed dose in order to avoid possible hematological effects.
Carcinogenesis, mutagenesis, impairment of fertility: In male rats, a statistically significant increase in urinary bladder tumors (transitional cell papilloma and carcinoma) was found only in association with xanthine calculi in the high dose group, at approximately 11 times human exposure. There was no significant increase in any other tumor type in either male or female mice or rats. These findings are considered a consequence of species-specific purine metabolism and urine composition and of no relevance to clinical use. A standard battery of test for genotoxicity did not reveal any biologically relevant genotoxic effects for Febuxostat. Febuxostat at oral doses up to 48 mg/kg/day was found to have no effect on fertility and reproductive performance of male and female rats. There was no evidence of impaired fertility, teratogenic effects, or harm to the fetus due to Febuxostat. There was high dose maternal toxicity accompanied by a reduction in weaning index and reduced development of offspring in rats at approximately 4.3 times human exposure. Teratology studies, performed in pregnant rats at approximately 4.3 times and pregnant rabbits at approximately 13 times human exposure did not reveal any teratogenic effects.