


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of Action: RANKL exists as a transmembrane or soluble protein. RANKL is essential for the formation, function and survival of osteoclasts, the sole cell type responsible for bone resorption. Increased osteoclast activity, stimulated by RANKL, is a key mediator of bone destruction in metastatic bone disease and multiple myeloma. Denosumab is a human monoclonal antibody (IgG2) that targets and binds with high affinity and specificity to RANKL, preventing the RANKL/RANK interaction from occurring and resulting in reduced osteoclast numbers and function, thereby decreasing bone resorption and cancer-induced bone destruction.
Giant cell tumours of bone are characterised by neoplastic stromal cells expressing RANK ligand and osteoclast-like giant cells expressing RANK. In patients with giant cell tumour of bone, denosumab binds to RANK ligand, significantly reducing or eliminating osteoclast-like giant cells. Consequently, osteolysis is reduced and proliferative tumour stroma is replaced with non-proliferative, differentiated, densely woven new bone.
Pharmacodynamic effects: In phase II clinical studies of patients with advanced malignancies involving bone, subcutaneous (SC) dosing of XGEVA administered either every 4 weeks (Q4W) or every 12 weeks resulted in a rapid reduction in markers of bone resorption (uNTX/Cr, serum CTx) with median reductions of approximately 80% for uNTX/Cr occurring within 1 week regardless of prior bisphosphonate therapy or baseline uNTx/Cr level. In phase III clinical trials of patients with advanced malignancies involving bone, median uNTx/Cr reductions of approximately 80% were maintained through 49 weeks of XGEVA treatment (120 mg every Q4W).
Immunogenicity: In clinical studies, neutralising antibodies have not been observed for denosumab in advanced cancer patients or giant cell tumour of bone patients. Using a sensitive immunoassay < 1% of patients treated with denosumab for up to 3 years tested positive for non-neutralising binding antibodies with no evidence of altered pharmacokinetics, toxicity, or clinical response.
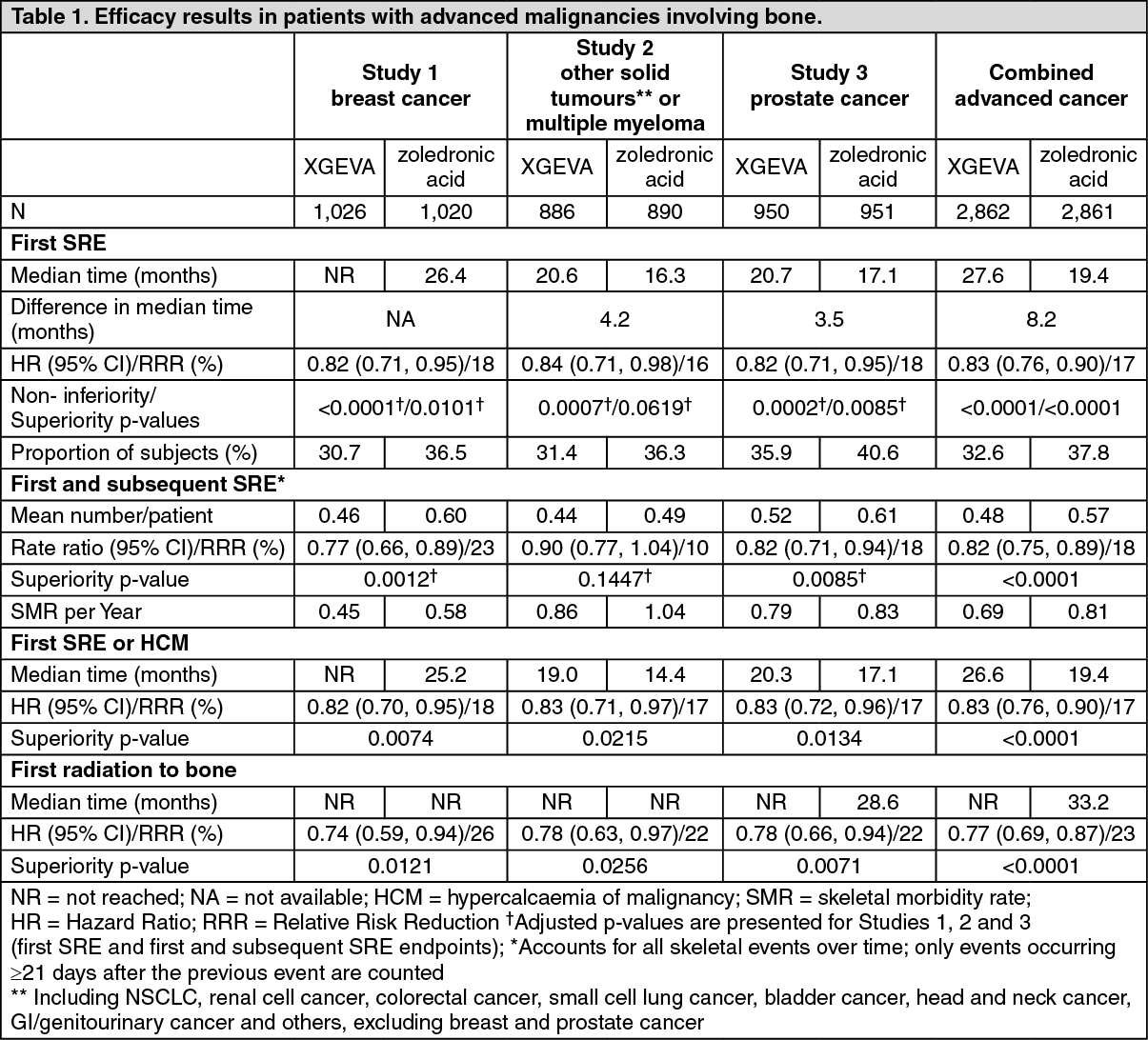
Clinical efficacy and safety in patients with bone metastases from solid tumours: Efficacy and safety of 120 mg XGEVA SC every 4 weeks or 4 mg zoledronic acid (dose-adjusted for reduced renal function) IV every 4 weeks were compared in three randomised, double-blind, active-controlled studies, in IV-bisphosphonate naïve patients with advanced malignancies involving bone: adults with breast cancer (study 1), other solid tumours or multiple myeloma (study 2), and castrate-resistant prostate cancer (study 3). Within these active-controlled clinical trials, safety was evaluated in 5,931 patients. Patients with prior history of ONJ or osteomyelitis of the jaw, an active dental or jaw condition requiring oral surgery, non-healed dental/oral surgery, or any planned invasive dental procedure, were not eligible for inclusion in these studies. The primary and secondary endpoints evaluated the occurrence of one or more skeletal-related events (SREs). In studies demonstrating superiority of XGEVA to zoledronic acid, patients were offered open-label XGEVA in a pre-specified 2-year extension treatment phase. An SRE was defined as any of the following: pathologic fracture (vertebral or non-vertebral), radiation therapy to bone (including the use of radioisotopes), surgery to bone, or spinal cord compression.
XGEVA reduced the risk of developing a SRE, and developing multiple SREs (first and subsequent) in patients with bone metastases from solid tumours (see Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image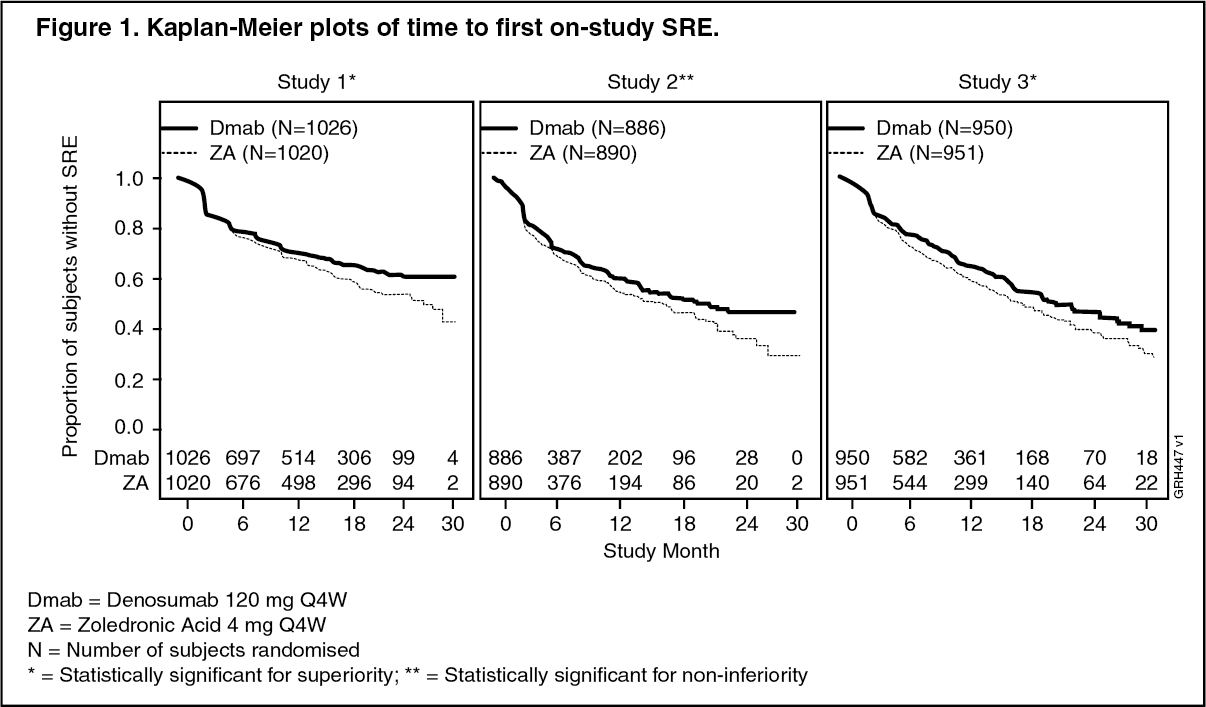 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDisease progression and overall survival with bone metastases from solid tumours: Disease progression was similar between XGEVA and zoledronic acid in all three studies and in the pre-specified analysis of all three studies combined.
In studies 1, 2 and 3, overall survival was balanced between XGEVA and zoledronic acid in patients with advanced malignancies involving bone: patients with breast cancer (hazard ratio and 95% CI was 0.95 [0.81, 1.11]), patients with prostate cancer (hazard ratio and 95% CI was 1.03 [0.91, 1.17]), and patients with other solid tumours or multiple myeloma (hazard ratio and 95% CI was 0.95 [0.83, 1.08]). A post-hoc analysis in study 2 (patients with other solid tumours or multiple myeloma) examined overall survival for the 3 tumour types used for stratification (non-small cell lung cancer, multiple myeloma, and other). Overall survival was longer for XGEVA in non-small cell lung cancer (hazard ratio [95% CI] of 0.79 [0.65, 0.95]; n = 702) and longer for zoledronic acid in multiple myeloma (hazard ratio [95% CI] of 2.26 [1.13, 4.50]; n = 180) and similar between XGEVA and zoledronic acid in other tumour types (hazard ratio [95% CI] of 1.08 (0.90, 1.30); n = 894). This study did not control for prognostic factors and anti-neoplastic treatments. In a combined pre-specified analysis from studies 1, 2 and 3, overall survival was similar between XGEVA and zoledronic acid (hazard ratio and 95% CI 0.99 [0.91, 1.07]).
Effect on pain: The time to pain improvement (i.e., ≥ 2-point decrease from baseline in BPI-SF worst pain score) was similar for denosumab and zoledronic acid in each study and the integrated analyses. In a post-hoc analysis of the combined dataset, the median time to worsening pain (> 4-point worst pain score) in patients with mild or no pain at baseline was delayed for XGEVA compared to zoledronic acid (198 versus 143 days) (p = 0.0002).
Clinical efficacy in patients with multiple myeloma: XGEVA was evaluated in an international, randomised (1:1), double-blind, active-controlled study comparing XGEVA with zoledronic acid in patients with newly diagnosed multiple myeloma, study 4.
In this study, 1,718 multiple myeloma patients with at least one bone lesion were randomised to receive 120 mg XGEVA subcutaneously every 4 weeks (Q4W) or 4 mg zoledronic acid intravenously (IV) every 4 weeks (dose-adjusted for renal function). The primary outcome measure was demonstration of non-inferiority of time to first on study skeletal-related event (SRE) as compared to zoledronic acid. Secondary outcome measures included superiority of time to first SRE, superiority of time to first and subsequent SRE, and overall survival. An SRE was defined as any of the following: pathologic fracture (vertebral or non-vertebral), radiation therapy to bone (including the use of radioisotopes), surgery to bone, or spinal cord compression.
Across both study arms, 54.5% of patients intended to undergo autologous PBSC transplantation, 95.8% patients utilised/planned to utilise a novel anti-myeloma agent (novel therapies include bortezomib, lenalidomide, or thalidomide) in first-line therapy, and 60.7% of patients had a previous SRE. The number of patients across both study arms with ISS stage I, stage II, and stage III at diagnosis were 32.4%, 38.2%, and 29.3%, respectively.
The median number of doses administered was 16 for XGEVA and 15 for zoledronic acid.
Efficacy results from study 4 are presented in figure 2 and table 2. (See Figure 2 and Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageClinical efficacy and safety in adults and skeletally mature adolescents with giant cell tumour of bone: The safety and efficacy of XGEVA was studied in two phase II open-label, single arm trials (studies 5 and 6) that enrolled 554 patients with giant cell tumour of bone that was either unresectable or for which surgery would be associated with severe morbidity. Patients received 120 mg XGEVA subcutaneously every 4 weeks with a loading dose of 120 mg on days 8 and 15. Patients who discontinued XGEVA then entered the safety follow-up phase for a minimum of 60 months.
Retreatment with XGEVA while in safety follow-up was allowed for subjects who initially demonstrated a response to XGEVA (e.g. in the case of recurrent disease).
Study 5 enrolled 37 adult patients with histologically confirmed unresectable or recurrent giant cell tumour of bone. The main outcome measure of the trial was response rate, defined as either at least 90% elimination of giant cells relative to baseline (or complete elimination of giant cells in cases where giant cells represent < 5% of tumour cells), or a lack of progression of the target lesion by radiographic measurements in cases where histopathology was not available. Of the 35 patients included in the efficacy analysis, 85.7% (95% CI: 69.7, 95.2) had a treatment response to XGEVA. All 20 patients (100%) with histology assessments met response criteria. Of the remaining 15 patients, 10 (67%) radiographic measurements showed no progression of the target lesion.
Study 6 enrolled 535 adult or skeletally mature adolescents with giant cell tumour of bone. Of these patients, 28 were aged 12-17 years. Patients were assigned to one of three cohorts: cohort 1 included patients with surgically unsalvageable disease (e.g. sacral, spinal, or multiple lesions, including pulmonary metastases); cohort 2 included patients with surgically salvageable disease whose planned surgery was associated with severe morbidity (e.g. joint resection, limb amputation, or hemipelvectomy); cohort 3 included patients previously participating in study 5 and rolled over into this study. The primary objective was to evaluate the safety profile of denosumab in subjects with giant cell tumour of bone. The secondary outcome measures of the study included time to disease progression (based on investigator assessment) for cohort 1 and proportion of patients without any surgery at month 6 for cohort 2.
In cohort 1 at the final analysis, 28 of the 260 treated patients (10.8%) had disease progression. In cohort 2, 219 of the 238 (92.0%; 95% CI: 87.8%, 95.1%) evaluable patients treated with XGEVA had not undergone surgery by month 6. Of the 239 subjects in cohort 2 with baseline target lesion location or on-study location not in lungs or soft tissue, a total of 82 subjects (34.3%) were able to avoid on-study surgery. Overall, efficacy results in skeletally mature adolescents were similar to those observed in adults.
Effect on pain: In the final analysis cohorts 1 and 2 combined, a clinically meaningful reduction in worst pain (i.e., ≥ 2 point decrease from baseline) was reported for 30.8% of patients at risk (i.e. those who had a worst pain score of ≥ 2 at baseline) within 1 week of treatment, and ≥ 50% at week 5. These pain improvements were maintained at all subsequent evaluations.
Paediatric population: In Study 6, XGEVA has been evaluated in a subset of 28 adolescent patients (aged 13-17 years) with giant cell tumour of bone who had reached skeletal maturity defined by at least 1 mature long bone (e.g. closed epiphyseal growth plate of the humerus) and body weight ≥ 45 kg. One adolescent subject with surgically unsalvageable disease (N=14) had disease recurrence during initial treatment. Thirteen of the 14 subjects with surgically salvageable disease whose planned surgery was associated with severe morbidity had not undergone surgery by month 6.
Pharmacokinetics: Absorption: Following subcutaneous administration, bioavailability was 62%.
Biotransformation: Denosumab is composed solely of amino acids and carbohydrates as native immunoglobulin and is unlikely to be eliminated via hepatic metabolic mechanisms. Its metabolism and elimination are expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides and individual amino acids.
Elimination: In subjects with advanced cancer, who received multiple doses of 120 mg every 4 weeks an approximate 2-fold accumulation in serum denosumab concentrations was observed and steady-state was achieved by 6 months, consistent with time-independent pharmacokinetics. In subjects with multiple myeloma who received 120 mg every 4 weeks, median trough levels varied by less than 8% between months 6 and 12. In subjects with giant cell tumour of bone who received 120 mg every 4 weeks with a loading dose on days 8 and 15, steady-state levels were achieved within the first month of treatment. Between weeks 9 and 49, median trough levels varied by less than 9%. In subjects who discontinued 120 mg every 4 weeks, the mean half-life was 28 days (range 14 to 55 days).
A population pharmacokinetic analysis did not indicate clinically significant changes in the systemic exposure of denosumab at steady-state with respect to age (18 to 87 years), race/ethnicity (Blacks, Hispanics, Asians and Caucasians explored), gender or solid tumour types or patients with multiple myeloma. Increasing body weight was associated with decreases in systemic exposure, and vice versa. The alterations were not considered clinically relevant, since pharmacodynamic effects based on bone turnover markers were consistent across a wide range of body weight.
Linearity/non-linearity: Denosumab displayed non-linear pharmacokinetics with dose over a wide dose range, but approximately dose-proportional increases in exposure for doses of 60 mg (or 1 mg/kg) and higher. The non-linearity is likely due to a saturable target-mediated elimination pathway of importance at low concentrations.
Renal impairment: In studies of denosumab (60 mg, n = 55 and 120 mg, n = 32) in patients without advanced cancer but with varying degrees of renal function, including patients on dialysis, the degree of renal impairment had no effect on the pharmacokinetics of denosumab; thus dose adjustment for renal impairment is not required. There is no need for renal monitoring with XGEVA dosing.
Hepatic impairment: No specific study in patients with hepatic impairment was performed. In general, monoclonal antibodies are not eliminated via hepatic metabolic mechanisms. The pharmacokinetics of denosumab is not expected to be affected by hepatic impairment.
Elderly: No overall differences in safety or efficacy were observed between geriatric patients and younger patients. Controlled clinical studies of XGEVA in patients with advanced malignancies involving bone over age 65 revealed similar efficacy and safety in older and younger patients. No dose adjustment is required in elderly patients.
Paediatric population: In skeletally mature adolescents (12-17 years of age) with giant cell tumour of bone who received 120 mg every 4 weeks with a loading dose on days 8 and 15, the pharmacokinetics of denosumab were similar to those observed in adult subjects with GCTB.
Toxicology: Preclinical safety data: Since the biological activity of denosumab in animals is specific to nonhuman primates, evaluation of genetically engineered (knockout) mice or use of other biological inhibitors of the RANK/RANKL pathway, such as OPG-Fc and RANK-Fc, were used to evaluate the pharmacodynamic properties of denosumab in rodent models.
In mouse bone metastasis models of oestrogen receptor positive and negative human breast cancer, prostate cancer and non-small cell lung cancer, OPG-Fc reduced osteolytic, osteoblastic, and osteolytic/osteoblastic lesions, delayed formation of de novo bone metastases, and reduced skeletal tumour growth. When OPG-Fc was combined with hormonal therapy (tamoxifen) or chemotherapy (docetaxel) in these models, there was additive inhibition of skeletal tumour growth in breast, and prostate or lung cancer respectively. In a mouse model of mammary tumour induction, RANK-Fc reduced hormone-induced proliferation in mammary epithelium and delayed tumour formation.
Standard tests to investigate the genotoxicity potential of denosumab have not been evaluated, since such tests are not relevant for this molecule. However, due to its character it is unlikely that denosumab has any potential for genotoxicity.
The carcinogenic potential of denosumab has not been evaluated in long-term animal studies.
In single and repeated dose toxicity studies in cynomolgus monkeys, denosumab doses resulting in 2.7 to 15 times greater systemic exposure than the recommended human dose had no impact on cardiovascular physiology, male or female fertility, or produced specific target organ toxicity.
In a study of cynomolgus monkeys dosed with denosumab during the period equivalent to the first trimester of pregnancy, denosumab doses resulting in 9 times greater systemic exposure than the recommended human dose did not induce maternal toxicity or foetal harm during a period equivalent to the first trimester, although foetal lymph nodes were not examined.
In another study of cynomolgus monkeys dosed with denosumab throughout pregnancy at systemic exposures 12-fold higher than the human dose, there were increased stillbirths and postnatal mortality; abnormal bone growth resulting in reduced bone strength, reduced haematopoiesis, and tooth malalignment; absence of peripheral lymph nodes; and decreased neonatal growth. A no observed adverse effect level for reproductive effects was not established. Following a 6 month period after birth, bone related changes showed recovery and there was no effect on tooth eruption. However, the effects on lymph nodes and tooth malalignment persisted, and minimal to moderate mineralisation in multiple tissues was seen in one animal (relation to treatment uncertain). There was no evidence of maternal harm prior to labour; adverse maternal effects occurred infrequently during labour. Maternal mammary gland development was normal.
In preclinical bone quality studies in monkeys on long-term denosumab treatment, decreases in bone turnover were associated with improvement in bone strength and normal bone histology.
In male mice genetically engineered to express huRANKL (knock-in mice), which were subjected to a transcortical fracture, denosumab delayed the removal of cartilage and remodelling of the fracture callus compared to control, but biomechanical strength was not adversely affected.
In preclinical studies knockout mice lacking RANK or RANKL had an absence of lactation due to inhibition of mammary gland maturation (lobulo-alveolar gland development during pregnancy) and exhibited impairment of lymph node formation. Neonatal RANK/RANKL knockout mice exhibited decreased body weight, reduced bone growth, altered growth plates and lack of tooth eruption. Reduced bone growth altered growth plates and impaired tooth eruption were also seen in studies of neonatal rats administered RANKL inhibitors, and these changes were partially reversible when dosing of RANKL inhibitor was discontinued. Adolescent primates dosed with denosumab at 2.7 and 15 times (10 and 50 mg/kg dose) the clinical exposure had abnormal growth plates. Therefore, treatment with denosumab may impair bone growth in children with open growth plates and may inhibit eruption of dentition.