The information highlighted (if any) are the most recent updates for this brand.
Each film-coated tablet contains 25 mg of agomelatine.
Excipient with known effect: Each film-coated tablet contains 61.8 mg lactose (as monohydrate).
Excipients/Inactive Ingredients: Tablet core: Lactose monohydrate, Maize starch, Povidone (K30), Sodium starch glycolate type A, Stearic acid, Magnesium stearate, Colloidal anhydrous silica.
Film-coating: Hypromellose, Yellow iron oxide (E172), Glycerol, Macrogol 6000, Magnesium stearate, Titanium dioxide (E171).
Printing ink containing shellac, propylene glycol and indigo carmine aluminium lake (E132).
Pharmacotherapeutic group: Psychoanaleptics, other antidepressants. ATC-code: N06AX22.
Pharmacology: Pharmacodynamics: Mechanism of action: Agomelatine is a melatonergic agonist (MT1 and MT2 receptors) and 5-HT2C antagonist. Binding studies indicate that agomelatine has no effect on monoamine uptake and no affinity for α, β adrenergic, histaminergic, cholinergic, dopaminergic and benzodiazepine receptors.
Agomelatine resynchronises circadian rhythms in animal models of circadian rhythm disruption. Agomelatine increases noradrenaline and dopamine release specifically in the frontal cortex and has no influence on the extracellular levels of serotonin.
Pharmacodynamic effects: Agomelatine has shown an antidepressant-like effect in animal models of depression (learned helplessness test, despair test, chronic mild stress) as well as in models with circadian rhythm desynchronisation and in models related to stress and anxiety.
In humans, agomelatine has positive phase shifting properties; it induces a phase advance of sleep, body temperature decline and melatonin onset.
Clinical efficacy and safety: Major depressive episodes: The efficacy and safety of agomelatine in major depressive episodes have been studied in a clinical programme including 7,900 patients treated with agomelatine.
Ten placebo controlled trials have been performed to investigate the short term efficacy of agomelatine in major depressive disorder in adults, with fixed dose and/or dose up-titration. At the end of treatment (over 6 or 8 weeks), significant efficacy of agomelatine 25-50 mg was demonstrated in 6 out of the ten short-term double-blind placebo-controlled trials. Primary endpoint was change in HAMD-17 score from baseline. Agomelatine failed to differentiate from placebo in two trials where the active control, paroxetine or fluoxetine showed assay sensitivity. Agomelatine was not compared directly with paroxetine and fluoxetine as these comparators were added in order to ensure assay sensitivity of the trials. In two other trials, it was not possible to draw any conclusions because the active controls, paroxetine or fluoxetine, failed to differentiate from placebo. However, in these studies it was not allowed to increase the start dose of either agomelatine, paroxetine or fluoxetine even if the response was not adequate.
Efficacy was also observed in more severely depressed patients (baseline HAM-D ≥ 25) in all positive placebo-controlled trials.
Response rates were statistically significantly higher with agomelatine compared with placebo.
Superiority (2 trials) or non-inferiority (4 trials) has been shown in six out of seven efficacy trials in heterogeneous populations of depressed adult patients versus SSRI/SNRI (sertraline, escitalopram, fluoxetine, venlafaxine or duloxetine). The anti-depressive effect was assessed with the HAMD-17 score either as primary or secondary endpoint.
The maintenance of antidepressant efficacy was demonstrated in a relapse prevention trial. Patients responding to 8/10-weeks of acute treatment with open-label agomelatine 25-50 mg once daily were randomised to either agomelatine 25-50 mg once daily or placebo for further 6-months. Agomelatine 25-50 mg once daily demonstrated a statistically significant superiority compared to placebo (p=0.0001) on the primary outcome measure, the prevention of depressive relapse, as measured by time to relapse. The incidence of relapse during the 6-months double-blind follow up period was 22% and 47% for agomelatine and placebo, respectively.
A placebo-controlled 8-week trial of agomelatine 25-50mg/day in elderly depressed patients (≥65 years, N=222, of which 151 on agomelatine) demonstrated a statistically significant difference of 2.67 points on HAM-D total score, the primary outcome. Responder rate analysis favoured agomelatine. No improvement was observed in very elderly patients (≥75 years, N= 69, of which 48 on agomelatine). Tolerability of agomelatine in elderly patients was comparable to that seen in the younger adults.
A specific controlled, 3-week trial has been conducted in patients suffering from major depressive disorder and insufficiently improved with paroxetine (a SSRI) or venlafaxine (a SNRI). When treatment was switched from these antidepressants to agomelatine, discontinuation symptoms arose after cessation of the SSRI or SNRI treatment, either after abrupt cessation or gradual cessation of the previous treatment. These discontinuation symptoms may be confounded with a lack of early benefit of agomelatine.
The percentage of patients with at least one discontinuation symptom one week after the SSRI/SNRI treatment stop, was lower in the long tapering group (gradual cessation of the previous SSRI/SNRI within 2 weeks) than in the short tapering group (gradual cessation of the previous SSRI/SNRI within 1 week) and in the abrupt substitution group (abrupt cessation): 56.1%, 62.6 % and 79.8% respectively. In a trial designed to assess discontinuation symptoms by the Discontinuation Emergent Signs and Symptoms (DESS) check-list in patients with remitted depression, agomelatine did not induce discontinuation syndrome after abrupt treatment cessation.
Generalized anxiety disorder: The efficacy and safety of agomelatine (25 & 25-50mg/day) in generalised anxiety disorder have been studied in a clinical programme including more than 1,100 GAD patients treated with agomelatine. Agomelatine (25 & 25-50mg once daily) demonstrated statistically significant superiority over placebo as measured by improvement in the Hamilton Anxiety Scale (HAM-A) total score in three out of three short term (12-week treatment), randomised, double-blind, placebo-controlled studies in adult patients.
Response and remission rates were also higher with agomelatine compared to placebo.
Assay sensitivity was shown in the trial with the active control, escitalopram.
Efficacy was also observed in more severely anxious patients (baseline HAMA ≥ 25) in all placebo-controlled trials.
Superiority versus placebo on global functioning was demonstrated using the Sheehan Disability Scale (SDS) in 2 out of 3 short term studies.
Agomelatine efficacy was compared directly with escitalopram in one study in patients suffering from severe generalised anxiety disorder (W0 HAMA ≥ 25), using the HAMA as the primary endpoint. In this study, agomelatine showed similar efficacy results to escitalopram in terms of improvements on the HAM-A total score.
The maintenance of efficacy in generalised anxiety disorder was demonstrated in a relapse prevention trial. Patients responding to 16-weeks of acute treatment with open-label agomelatine 25mg once daily with a possible up titration to 50 mg (once daily) after 4 weeks were randomised to either agomelatine 25-50mg or placebo for further 6-months (26 weeks). Agomelatine 25-50 mg once daily demonstrated a statistically significant superiority compared to placebo (p=0.046) on the primary outcome measure, the prevention of anxious relapse, as measured by time to relapse. The incidence of relapse during the 6-months double-blind follow up period was 20% and 31% for agomelatine and placebo, respectively. In this study, discontinuation symptoms were assessed by the Discontinuation Emergent Signs and Symptoms (DESS) check-list in patients having completed the study up to week 42 and rerandomised either on placebo or agomelatine. Absence of discontinuation syndrome after abrupt agomelatine treatment cessation was confirmed in this population.
Efficacy in elderly GAD patients has not been assessed in a specific study, data from the performed studies are very limited thus, agomelatine should not be used in GAD patients over 65 years.
General properties: Agomelatine does not alter daytime vigilance and memory in healthy volunteers. In depressed patients, treatment with agomelatine 25 mg increased slow wave sleep without modification of REM (Rapid Eye Movement) sleep amount or REM latency. Agomelatine 25 mg also induced an advance of the time of sleep onset and of minimum heart rate. From the first week of treatment, onset of sleep and the quality of sleep were significantly improved without daytime clumsiness as assessed by patients.
In healthy volunteers agomelatine preserved sexual function in comparison with paroxetine. In a specific sexual dysfunction comparative trial with remitted depressed patients, there was a numerical trend (not statistically significant) towards less sexual emergent dysfunction than venlafaxine for Sex Effects Scale (SEXFX) drive arousal or orgasm scores on agomelatine. The pooled analysis of trials using the Arizona Sexual Experience Scale (ASEX) showed that agomelatine was not associated with sexual dysfunction.
Agomelatine had neutral effect on heart rate and blood pressure in clinical trials.
Agomelatine has no abuse potential as measured in healthy volunteer studies on a specific visual analogue scale or the Addiction Research Center Inventory (ARCI) 49 check-list.
Pharmacokinetics: Absorption and bioavailability: Agomelatine is rapidly and well (≥ 80%) absorbed after oral administration. Absolute bioavailability is low (< 5% at the therapeutic oral dose) and the interindividual variability is substantial. The bioavailability is increased in women compared to men. The bioavailability is increased by intake of oral contraceptives and reduced by smoking. The peak plasma concentration is reached within 1 to 2 hours.
In the therapeutic dose-range, agomelatine systemic exposure increases proportionally with dose. At higher doses, a saturation of the first-pass effect occurs.
Food intake (standard meal or high fat meal) does not modify the bioavailability or the absorption rate. The variability is increased with high fat food.
Distribution: Steady state volume of distribution is about 35 l and plasma protein binding is 95% irrespective of the concentration and is not modified with age and in patients with renal impairment but the free fraction is doubled in patients with hepatic impairment.
Biotransformation: Following oral administration, agomelatine is rapidly metabolised mainly via hepatic CYP1A2; CYP2C9 and CYP2C19 isoenzymes are also involved but with a low contribution.
The major metabolites, hydroxylated and demethylated agomelatine, are not active and are rapidly conjugated and eliminated in the urine.
Elimination: Elimination is rapid, the mean plasma half-life is between 1 and 2 hours and the clearance is high (about 1,100 ml/min) and essentially metabolic.
Excretion is mainly (80%) urinary and in the form of metabolites, whereas unchanged compound recovery in urine is negligible.
Kinetics are not modified after repeated administration.
Renal impairment: No relevant modification of pharmacokinetic parameters in patients with severe renal impairment has been observed (n=8, single dose of 25 mg), but caution should be exercised in patients with severe or moderate renal impairment as only limited clinical data are available in these patients (see Dosage & Administration).
Hepatic impairment: In a specific study involving cirrhotic patients with chronic mild (Child-Pugh type A) or moderate (Child-Pugh type B) liver impairment, exposure to agomelatine 25 mg was substantially increased (70-times and 140-times, respectively), compared to matched volunteers (age, weight and smoking habit) with no liver failure (see Dosage & Administration, Contraindications and Precautions).
Elderly: In a pharmacokinetic study in elderly patients (≥ 65 years), it was showed that at a dose of 25 mg the mean AUC and mean Cmax were about 4-fold and 13-fold higher for patients ≥ 75 years old compared to patients < 75 years old. The total number of patients receiving 50 mg was too low to draw any conclusions. No dose adaptation is required in elderly patients.
Ethnic groups: There is no data on the influence of race on agomelatine pharmacokinetics.
Toxicology: Preclinical safety data: In mice, rats and monkeys sedative effects were observed after single and repeated administration at high doses.
In rodents, a marked induction of CYP2B and a moderate induction of CYP1A and CYP3A were seen from 125 mg/kg/day whereas in monkeys the induction was slight for CYP2B and CYP3A at 375 mg/kg/day. No hepatotoxicity was observed in rodents and monkeys in the repeat dose toxicity studies.
Agomelatine passes into the placenta and foetuses of pregnant rats.
Reproduction studies in the rat and the rabbit showed no effect of agomelatine on fertility, embryofoetal development and pre- and post natal development.
A battery of in vitro and in vivo standard genotoxicity assays concludes to no mutagenic or clastogenic potential of agomelatine.
In carcinogenicity studies agomelatine induced an increase in the incidence of liver tumours in the rat and the mouse, at a dose at least 110-fold higher than the therapeutic dose. Liver tumours are most likely related to enzyme induction specific to rodents. The frequency of benign mammary fibroadenomas observed in the rat was increased with high exposures (60-fold the exposure at the therapeutic dose) but remains in the range of that of controls.
Safety pharmacology studies showed no effect of agomelatine on hERG (human Ether à-go-go Related Gene) current or on dog Purkinje cells action potential. Agomelatine did not show proconvulsive properties at ip doses up to 128 mg/kg in mice and rats.
No effect of agomelatine on juvenile animals behavioural performances, visual and reproductive function were observed. There were mild non dose dependent decreases in body weight related to the pharmacological properties and some minor effects on male reproductive tract without any impairment on reproductive performances.
Valdoxan is indicated for the treatment of: major depressive episodes in adults; generalised anxiety disorder (GAD).
Posology: The recommended dose is 25 mg once daily taken orally at bedtime.
If there is no improvement of symptoms, the dose may be increased to 50 mg once daily, i.e. two 25 mg tablets, taken together at bedtime: 2 weeks after treatment initiation in major depressive episode; 4 weeks after treatment initiation in generalised anxiety disorder.
Decision of dose increase has to be balanced with a higher risk of transaminases elevation. Any dose increase to 50 mg should be made on an individual patient benefit/risk basis and with strict respect of Liver Function Test monitoring.
Liver function tests should be performed in all patients before starting treatment. Treatment should not be initiated if transaminases exceed 3 X upper limit of normal (see Contraindications and Precautions).
During treatment transaminases should be monitored periodically after around three weeks, six weeks (end of acute phase), twelve weeks and twenty four weeks (end of maintenance phase) and thereafter when clinically indicated (see also Precautions). Treatment should be discontinued if transaminases exceed 3 X upper limit of normal (see Contraindications and Precautions).
When increasing the dosage, liver function tests should again be performed at the same frequency as when initiating treatment.
Treatment duration: Patients should be treated for a sufficient period of at least 6 months to ensure that they are free of symptoms.
Switching therapy from SSRI/SNRI antidepressant to agomelatine: Patients may experience discontinuation symptoms after cessation from an SSRI/SNRI antidepressant.
The package insert of the actual SSRI/SNRI should be consulted on how to withdraw the treatment to avoid this. Agomelatine can be started immediately while tapering the dosage of a SSRI/SNRI (see Pharmacology: Pharmacodynamics under Actions).
Treatment discontinuation: No dosage tapering is needed on treatment discontinuation.
Special populations: Elderly: The efficacy and safety of agomelatine (25 to 50 mg/day) have been established in elderly depressed patients (< 75 years). No dose adjustment is required in relation to age (see Pharmacology: Pharmacokinetics under Actions).
No effect is documented in depressed patients ≥75 years nor in elderly patient suffering from generalised anxiety disorder. Therefore agomelatine should not be used by patients in this age group (see Precautions and Pharmacology: Pharmacodynamics under Actions).
Renal impairment: No relevant modification in agomelatine pharmacokinetic parameters in patients with severe renal impairment has been observed. However, only limited clinical data on the use of agomelatine in depressed patients with severe or moderate renal impairment with major depressive episodes is available. Therefore, caution should be exercised when prescribing agomelatine to these patients.
Hepatic impairment: Agomelatine is contraindicated in patients with hepatic impairment (see Contraindications, Precautions and Pharmacology: Pharmacokinetics under Actions).
Paediatric population: The safety and efficacy of agomelatine in children from 2 years onwards have not been established.
No data are available (see Precautions).
There is no relevant use of agomelatine in children from birth to 2 years.
Method of administration: For oral use.
Valdoxan film-coated tablets may be taken with or without food.
Symptoms: There is limited experience with agomelatine overdose. Experience with agomelatine in overdose has indicated that epigastralgia, somnolence, fatigue, agitation, anxiety, tension, dizziness, cyanosis or malaise have been reported.
One person having ingested 2,450 mg agomelatine, recovered spontaneously without cardiovascular and biological abnormalities.
Management: No specific antidotes for agomelatine are known. Management of overdose should consist of treatment of clinical symptoms and routine monitoring. Medical follow-up in a specialised environment is recommended.
Hypersensitivity to the active substance or to any of the excipients listed in Description.
Hepatic impairment (i.e. cirrhosis or active liver disease) or transaminases exceeding 3 X upper limit of normal (see Dosage & Administration and Precautions).
Concomitant use of potent CYP1A2 inhibitors (e.g. fluvoxamine, ciprofloxacin) (see Interactions).
Monitoring of liver function: Cases of liver injury, including hepatic failure (few cases were exceptionally reported with fatal outcome or liver transplantation in patients with hepatic risk factors), elevations of liver enzymes exceeding 10 times upper limit of normal, hepatitis and jaundice have been reported in patients treated with agomelatine in the post-marketing setting (see Adverse Reactions). Most of them occurred during the first months of treatment. The pattern of liver damage is predominantly hepatocellular with increased serum transaminases which usually return to normal levels on cessation of agomelatine.
Caution should be exercised before starting treatment and close surveillance should be performed throughout the treatment period in all patients, especially if hepatic injury risk factors or concomitant medicinal products associated with risk of hepatic injury are present.
Before starting treatment: Treatment with Valdoxan should only be prescribed after careful consideration of benefit and risk in patients with hepatic injury risk factors e.g.: obesity/overweight/non-alcoholic fatty liver disease, diabetes; alcohol use disorder and/or substantial alcohol intake; and in patients receiving concomitant medicinal products associated with risk of hepatic injury.
Baseline liver function tests should be undertaken in all patients and treatment should not be initiated in patients with baseline values of ALT and/or AST >3 X upper limit of normal (see Contraindications). Caution should be exercised when Valdoxan is administered to patients with pretreatment elevated transaminases (> the upper limit of the normal ranges and ≤3 times the upper limit of the normal range).
Frequency of liver function tests: before starting treatment and then: after around 3 weeks, after around 6 weeks (end of acute phase), after around 12 and 24 weeks (end of maintenance phase), and thereafter when clinically indicated.
When increasing the dosage, liver function tests should again be performed at the same frequency as when initiating treatment.
Any patient who develops increased serum transaminases should have his/her liver function tests repeated within 48 hours.
During treatment period: Valdoxan treatment should be discontinued immediately if: patient develops symptoms or signs of potential liver injury (such as dark urine, light coloured stools, yellow skin/eyes, pain in the upper right belly, sustained new-onset and unexplained fatigue); the increase in serum transaminases exceeds 3 X upper limit of normal.
Following discontinuation of Valdoxan therapy liver function tests should be repeated until serum transaminases return to normal.
Bipolar disorder/mania/hypomania: Valdoxan should be used with caution in patients with a history of bipolar disorder, mania or hypomania and should be discontinued if a patient develops manic symptoms (see Adverse Reactions).
Suicide/suicidal thoughts: Depression is associated with an increased risk of suicidal thoughts, self harm and suicide (suicide-related events). This risk persists until significant remission occurs. As improvement may not occur during the first few weeks or more of treatment, patients should be closely monitored until such improvement occurs. It is general clinical experience that the risk of suicide may increase in the early stages of recovery.
Generalised anxiety disorder for which Valdoxan is prescribed can also be associated with an increased risk of suicide-related events. The same precautions observed when treating depressed patients should therefore be observed when treating patients with generalised anxiety disorder.
Patients with a history of suicide-related events or those exhibiting a significant degree of suicidal ideation prior to commencement of treatment are known to be at greater risk of suicidal thoughts or suicide attempts, and should receive careful monitoring during treatment. A meta-analysis of placebo-controlled clinical trials of antidepressants in adult patients with psychiatric disorders showed an increased risk of suicidal behaviour with antidepressants compared to placebo, in patients less than 25 years old.
Close supervision of patients and in particular those at high risk should accompany treatment especially in early treatment and following dose changes. Patients (and caregivers of patients) should be alerted to the need to monitor for any clinical worsening, suicidal behaviour or thoughts and unusual changes in behaviour and to seek medical advice immediately if these symptoms present.
Combination with CYP1A2 inhibitors (see Contraindications and Interactions): Caution should be exercised when prescribing Valdoxan with moderate CYP1A2 inhibitors (e.g. propranolol, enoxacin) which may result in increased exposure of agomelatine.
Lactose intolerance: Valdoxan contains lactose. Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Level of sodium: Valdoxan contains less than 1 mmol sodium (23 mg) per tablet, i.e. essentially 'sodium-free'.
Effects on ability to drive and use machines: Agomelatine has minor influence on the ability to drive and use machines.
Considering that dizziness and somnolence are common adverse reactions patients should be cautioned about their ability to drive or operate machines.
Use in Children: Valdoxan is not recommended in patients under 18 years of age since safety and efficacy of Valdoxan have not been established in this age group. In clinical trials among children and adolescents treated with other antidepressants, suicide-related behaviour (suicide attempt and suicidal thoughts), and hostility (predominantly aggression, oppositional behaviour and anger) were more frequently observed compared to those treated with placebo (see Dosage & Administration).
Use in the Elderly: No effect of agomelatine is documented in depressed patients ≥75 years nor in elderly patients suffering from generalised anxiety disorder. Therefore agomelatine should not be used in these patients (see also Dosage & Administration and Pharmacology: Pharmacodynamics under Actions).
Use in elderly with dementia: Valdoxan should not be used for the treatment of major depressive episodes or generalised anxiety disorder in elderly patients with dementia since the safety and efficacy of Valdoxan have not been established in these patients.
Pregnancy: There are no or limited amount of data (less than 300 pregnancy outcomes) from the use of agomelatine in pregnant women. Animal studies do not indicate direct or indirect harmful effects with respect to pregnancy, embryonal/foetal development, parturition or postnatal development (see Pharmacology: Toxicology: Preclinical safety data under Actions). As a precautionary measure, it is preferable to avoid the use of Valdoxan during pregnancy.
Breast-feeding: It is not known whether agomelatine/metabolites are excreted in human milk. Available pharmacodynamic/toxicological data in animals have shown excretion of agomelatine/metabolites in milk (see Pharmacology: Toxicology: Preclinical safety data under Actions). A risk to the newborns/infants cannot be excluded. A decision must be made whether to discontinue breast-feeding or to discontinue/abstain from Valdoxan therapy taking into account the benefit of breast feeding for the child and the benefit of therapy for the woman.
Fertility: Reproduction studies in the rat and the rabbit showed no effect of agomelatine on fertility (see Pharmacology: Toxicology: Preclinical safety data under Actions).
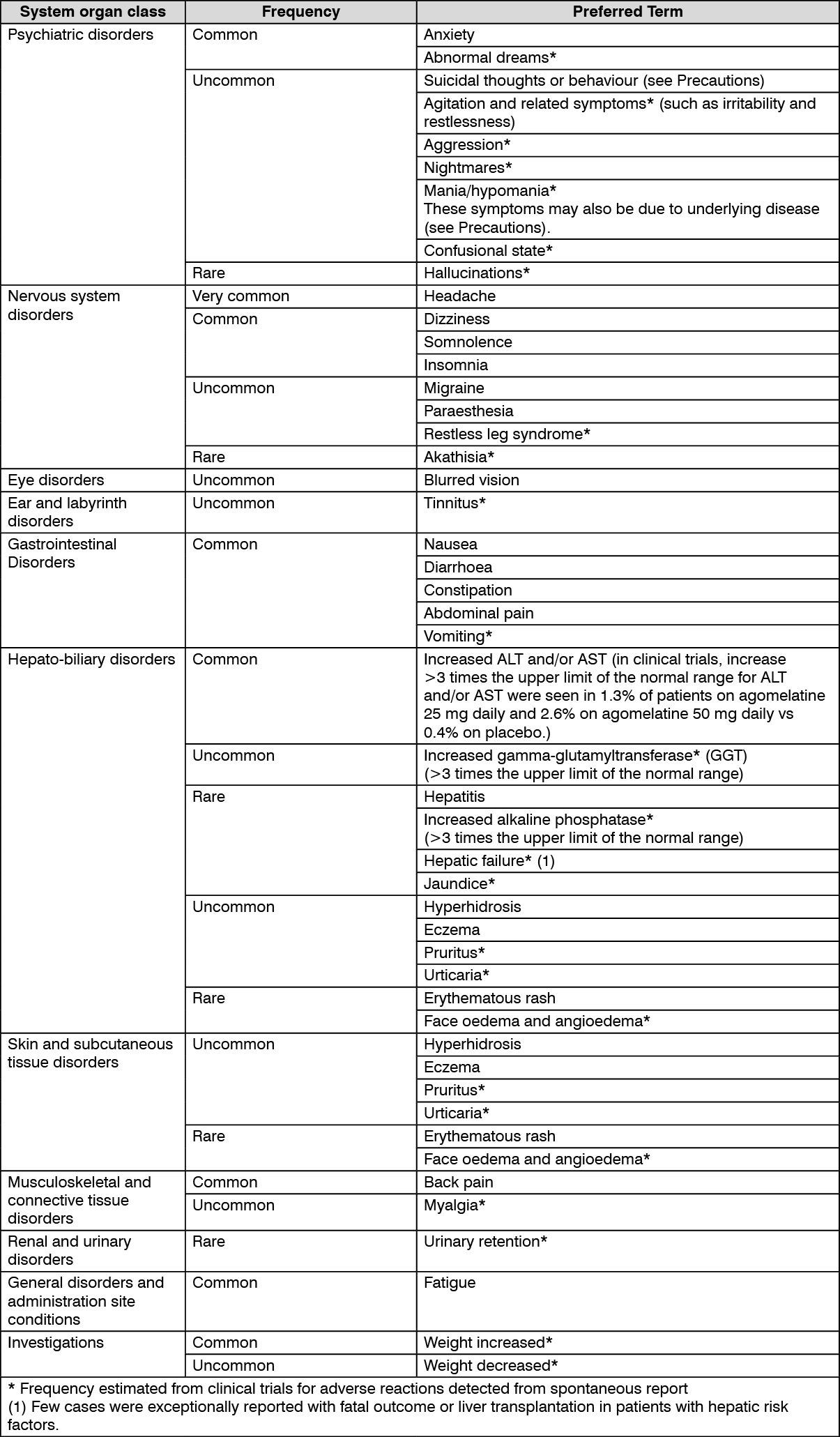
Summary of the safety profile: Adverse reactions were usually mild or moderate and occurred within the first two weeks of treatment. The most common adverse reactions were headache, nausea and dizziness.
These adverse reactions were usually transient and did not generally lead to cessation of therapy.
Tabulated list of adverse reactions: The following table gives the adverse reactions observed clinical trials
and post marketing spontaneous reports.
Adverse reactions are listed as follows using the following convention: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000), not known (cannot be estimated from the available data). The frequencies have not been corrected for placebo. (
See table.)
 Click on icon to see table/diagram/image
Reporting of suspected adverse reactions:
Click on icon to see table/diagram/image
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system.
Potential interactions affecting agomelatine: Agomelatine is metabolised mainly by cytochrome P450 1A2 (CYP1A2) (90%) and by CYP2C9/19 (10%). Medicinal products that interact with these isoenzymes may decrease or increase the bioavailability of agomelatine.
Fluvoxamine, a potent CYP1A2 and moderate CYP2C9 inhibitor markedly inhibits the metabolism of agomelatine resulting in a 60-fold (range 12-412) increase of agomelatine exposure.
Consequently, co-administration of Valdoxan with potent CYP1A2 inhibitors (e.g. fluvoxamine, ciprofloxacin) is contraindicated.
Combination of agomelatine with oestrogens (moderate CYP1A2 inhibitors) results in a several fold increased exposure of agomelatine. While there was no specific safety signal in the 800 patients treated in combination with oestrogens, caution should be exercised when prescribing agomelatine with other moderate CYP1A2 inhibitors (e.g. propranolol, enoxacin) until more experience has been gained (see Precautions).
Rifampicin an inducer of all three cytochromes involved in the metabolism of agomelatine may decrease the bioavailability of agomelatine.
Smoking induces CYP1A2 and has been shown to decrease the bioavailability of agomelatine, especially in heavy smokers (≥ 15 cigarettes/day) (see Pharmacology: Pharmacokinetics under Actions).
Potential for agomelatine to affect other medicinal products: In vivo, agomelatine does not induce CYP450 isoenzymes. Agomelatine inhibits neither CYP1A2 in vivo nor the other CYP450 in vitro. Therefore, agomelatine will not modify exposure to medicinal products metabolised by CYP 450.
Other medicinal products: No evidence of pharmacokinetic or pharmacodynamic interaction with medicinal products which could be prescribed concomitantly with Valdoxan in the target population was found in phase I clinical trials: benzodiazepines, lithium, paroxetine, fluconazole and theophylline.
Alcohol: The combination of agomelatine and alcohol is not advisable.
Electroconvulsive therapy (ECT): There is no experience of concurrent use of agomelatine with ECT. Animal studies have not shown proconvulsant properties (see Pharmacology: Toxicology: Preclinical safety data under Actions). Therefore, clinical consequences of ECT performed concomitantly with agomelatine treatment are considered to be unlikely.
Paediatric population: Interaction studies have only been performed in adults.
Incompatibilities: Not applicable.
Special precautions for disposal: No special requirements for disposal.
Store below 30°C.
Shelf life: 3 years.
N06AX22 - agomelatine ; Belongs to the class of other antidepressants.
Valdoxan FC tab 25 mg
28's




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
