Pharmacology: Mechanism of Action: Lapatinib is a 4-anilinoquinazoline kinase inhibitor of the intracellular tyrosine kinase domains of both Epidermal Growth Factor Receptor (EGFR [ErbB1]) and of Human Epidermal Receptor Type 2 (HER2 [ErbB2]) receptors (estimated K
iapp values of 3nM and 13 nM, respectively) with a dissociation half-life of greater than or equal to 300 minutes. Lapatinib inhibits ErbB-driven tumor cell growth in vitro and in various animal models.
An additive effect was demonstrated in an in vitro study when lapatinib and 5-FU (the active metabolite of capecitabine) were used in combination in the 4-tumor cell lines tested. The growth inhibitory effects of lapatinib were evaluated in trastuzumab-conditioned cell lines. Lapatinib retained significant activity against breast cancer cell lines selected for long-term growth in trastuzumab-containing medium in vitro. These in vitro findings suggest non-cross-resistance between these two agents.
Hormone receptor-positive breast cancer cells (with ER [Estrogen Receptor] and/or PgR [Progesterone Receptor]) that coexpress the HER2 tend to be resistant to established endocrine therapies. Similarly, hormone receptor-positive breast cancer cells that initially lack EGFR or HER2 upregulate these receptor proteins as the tumor becomes resistant to endocrine therapy.
Pharmacodynamics: Cardiac Electrophysiology: The effect of lapatinib on the QT-interval was evaluated in a single-blind, placebo-controlled, single sequence (placebo and active treatment) crossover study in patients with advanced solid tumors (N = 58). During the 4-day treatment period, three doses of matching placebo were administered 12 hours apart in the morning and evening on Day 1 and in the morning on Day 2. This was followed by three doses of lapatinib 2,000 mg (1.3 - 1.6 times the recommended dosage) administered in the same way. Measurements, including ECGs and pharmacokinetic samples were done at baseline and at the same time points on Day 2 and Day 4. In the evaluable population of subjects who had complete dosing and ECG assessments (N = 37), the maximum mean △△QTcF (90% CI) of 8.75 ms (4.08, 13.42) was observed 10 hours after ingestion of the third dose of lapatinib 2000 mg. The △△QTcF exceeded the 5 ms threshold and the upper bound 90% CIs exceeded the 10 ms threshold at multiple time points.
There was a concentration-dependent increase in QTcF effects [see QT Prolongation under Precautions].
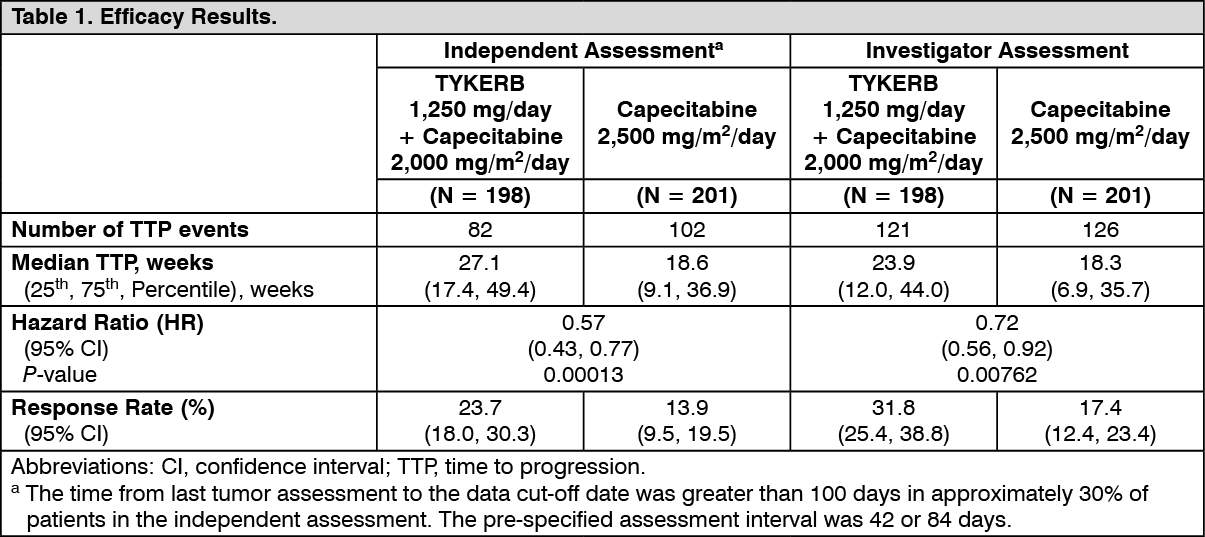
Clinical Studies: HER2-Positive Metastatic Breast Cancer: The efficacy and safety of TYKERB in combination with capecitabine in breast cancer were evaluated in a randomized, Phase 3 trial. Patients eligible for enrollment had HER2 (ErbB2) overexpressing (IHC 3+ or IHC 2+ confirmed by FISH), locally advanced or metastatic breast cancer, progressing after prior treatment that included anthracyclines, taxanes, and trastuzumab.
Patients were randomized to receive either TYKERB 1,250 mg once daily (continuously) plus capecitabine 2,000 mg/m
2/day on Days 1-14 every 21 days, or to receive capecitabine alone at a dose of 2,500 mg/m
2/day on Days 1-14 every 21 days. The endpoint was time to progression (TTP). TTP was defined as time from randomization to tumor progression or death related to breast cancer. Based on the results of a pre-specified interim analysis, further enrollment was discontinued. Three hundred and ninety-nine (399) patients were enrolled in this study. The median age was 53 years and 14% were older than 65 years. Ninety-one percent (91%) were Caucasian. Ninety-seven percent (97%) had stage IV breast cancer, 48% were estrogen receptor+ (ER+) or progesterone receptor+ (PR+), and 95% were ErbB2 IHC 3+ or IHC 2+ with FISH confirmation. Approximately 95% of patients had prior treatment with anthracyclines, taxanes, and trastuzumab.
Efficacy analyses 4 months after the interim analysis are presented in Table 1, Figures 1, and 2. (See Table 1 and Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
At the time of previously mentioned efficacy analysis, the overall survival (OS) data were not mature (32% events). However, based on the TTP results, the study was unblinded and patients receiving capecitabine alone were allowed to cross over to treatment with TYKERB plus capecitabine. The survival data were followed for an additional 2 years to be mature and the analysis is summarized in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
Clinical Studies Describing Limitations of Use: In two randomized trials, TYKERB-based chemotherapy regimens have been shown to be less effective than trastuzumab-based chemotherapy regimens. The first randomized, open-label study compared the safety and efficacy of TYKERB in combination with capecitabine relative to trastuzumab in combination with capecitabine in women with HER2-positive metastatic breast cancer (N = 540). The study was stopped early based on the findings of a pre-planned interim analysis showing a low incidence of CNS events (primary endpoint) and superior efficacy of the trastuzumab plus capecitabine. The median progression-free survival was 6.6 months in the group receiving TYKERB in combination with capecitabine compared with 8.0 months in the group receiving the trastuzumab combination [HR = 1.30 (95% CI: 1.04, 1.64)]. Overall survival was analyzed when 26% of deaths occurred in the group receiving TYKERB in combination with capecitabine and 22% in the group receiving the trastuzumab combination [HR = 1.34 (95% CI: 0.95, 1.92)].
The second randomized, open-label study compared the safety and efficacy of taxane-based chemotherapy plus TYKERB to taxane-based chemotherapy plus trastuzumab as first-line therapy in women with HER2-positive, metastatic breast cancer (N = 652). The study was stopped early based on findings from a pre-planned interim analysis. The median progression-free survival was 11.3 months in the trastuzumab combination treatment arm compared to 9.0 months in patients treated with TYKERB in the combination arm for the intent-to-treat population [HR = 1.37 (95% CI: 1.13, 1.65)].
Hormone Receptor-Positive, HER2-Positive Metastatic Breast Cancer: The efficacy and safety of TYKERB in combination with letrozole were evaluated in a double-blind, placebo-controlled, multi-center study. A total of 1286 postmenopausal women with hormone receptor-positive (ER positive and/or PgR positive) metastatic breast cancer, who had not received prior therapy for metastatic disease, were randomly assigned to receive either TYKERB (1,500 mg once daily) plus letrozole (2.5 mg once daily) (n = 642) or letrozole (2.5 mg once daily) alone (n = 644). Of all patients randomized to treatment, 219 (17%) patients had tumors overexpressing the HER2 receptor, defined as fluorescence in situ hybridization (FISH) greater than or equal to 2 or 3+ immunohistochemistry (IHC). There were 952 (74%) patients who were HER2-negative and 115 (9%) patients did not have their HER2 receptor status confirmed. The primary objective was to evaluate and compare progression-free survival (PFS) in the HER2-positive population. Progression-free survival was defined as the interval of time between date of randomization and the earlier date of first documented sign of disease progression or death due to any cause.
The baseline demographic and disease characteristics were balanced between the two treatment arms. The median age was 63 years and 45% were 65 years of age or older. Eighty-four percent (84%) of the patients were white. Approximately 50% of the HER2-positive population had prior adjuvant/neo-adjuvant chemotherapy and 56% had prior hormonal therapy. Only 2 patients had prior trastuzumab.
In the HER2-positive subgroup (n = 219), the addition of TYKERB to letrozole resulted in an improvement in PFS. In the HER2-negative subgroup, there was no improvement in PFS of the combination of TYKERB plus letrozole compared to the letrozole plus placebo. Overall response rate (ORR) was also improved with the combination of TYKERB plus letrozole. The OS data were not mature. Efficacy analyses for the hormone receptor-positive, HER2-positive and HER2-negative subgroups are presented in Table 3 and Figure 3. (See Table 3 and Figure 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The efficacy and safety of TYKERB, in combination with an aromatase inhibitor (AI), were confirmed in another randomized Phase 3 trial. Patients enrolled were post-menopausal women who had hormone receptor-positive (HR+)/HER2-positive, metastatic breast cancer, which had progressed after prior trastuzumab-containing chemotherapy and endocrine therapies. A total of 355 patients were randomized in a 1:1:1 ratio to TYKERB 1000 mg + trastuzumab + AI (N = 120), or trastuzumab + AI arm (N = 117), or TYKERB 1500 mg + AI (N = 118). In the trastuzumab-containing arms, trastuzumab was administered with a loading dose of 8 mg/kg IV followed by the maintenance dose of 6 mg/kg every 3 weeks. In the AI-containing arms, the AIs were administered at doses of letrozole 2.5 mg once daily, or exemestane 25 mg once daily, or anastrozole 1 mg once daily.
The study was designed to evaluate a potential benefit in Progression Free Survival (PFS) when double versus single HER2 targeted therapy was administered in combination with an AI (letrozole, exemestane, or anastrozole). The major efficacy outcome measure was PFS based on local radiology/investigator's assessment comparing TYKERB + trastuzumab + AI versus trastuzumab + AI.
The median age was 56 years (range 30 to 84). The majority of the patients treated on this trial (70%) were Caucasian.
Efficacy results are presented in Table 4 and Figure 4. The OS data were not mature (23% of patients had died). (See Table 4 and Figure 4.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacokinetics: Absorption: Absorption following oral administration of TYKERB is incomplete and variable. Serum concentrations appear after a median lag time of 0.25 hours (range 0 to 1.5 hours). Peak plasma concentrations (C
max) of lapatinib are achieved approximately 4 hours after administration. Daily dosing of TYKERB results in the achievement of steady-state within 6 to 7 days, indicating an effective half-life of 24 hours.
At the dose of 1,250 mg daily, steady-state geometric mean [95% confidence interval (CI)] values of C
max were 2.43 mcg/mL (1.57 to 3.77 mcg/mL) and AUC were 36.2 mcg·h/mL (23.4 to 56 mcg·h/mL).
Divided daily doses of TYKERB resulted in approximately 2-fold higher exposure at steady-state (steady-state AUC) compared to the same total dose administered once daily.
Systemic exposure to lapatinib is increased when administered with food. Lapatinib AUC values were approximately 3- and 4-fold higher (C
max approximately 2.5- and 3-fold higher) when administered with a low-fat (5% fat-500 calories) or with a high-fat (50% fat-1000 calories) meal, respectively.
Distribution: Lapatinib is highly bound (greater than 99%) to albumin and alpha-1 acid glycoprotein. In vitro studies indicate that lapatinib is a substrate for the transporters of breast cancer-resistance protein (BCRP, ABCG2) and P-glycoprotein (P-gp, ABCB1). Lapatinib has also been shown to inhibit P-gp, BCRP, and the hepatic uptake transporter OATP 1B1, in vitro at clinically relevant concentrations.
Metabolism: Lapatinib undergoes extensive metabolism, primarily by CYP3A4 and CYP3A5, with minor contributions from CYP2C19 and CYP2C8 to a variety of oxidated metabolites, none of which accounts for more than 14% of the dose recovered in the feces or 10% of lapatinib concentration in the plasma.
Elimination: At clinical doses, the terminal phase half-life following a single dose was 14.2 hours; accumulation with repeated dosing indicates an effective half-life of 24 hours.
Elimination of lapatinib is predominantly through metabolism by CYP3A4/5 with negligible (less than 2%) renal excretion. Recovery of parent lapatinib in feces accounts for a median of 27% (range 3% to 67%) of an oral dose.
Effects of Age, Gender, or Race: Studies of the effects of age, gender, or race on the pharmacokinetics of lapatinib have not been performed.
Pharmacogenomics: The HLA alleles DQA1*02:01 and DRB1*07:01 were associated with hepatotoxicity reactions in a genetic substudy of a monotherapy trial with TYKERB (n = 1194). Severe liver injury (ALT greater than 5 times the upper limit of normal, NCI CTCAE Grade 3) occurred in 2% of patients overall; the incidence of severe liver injury among DQA1*02:01 or DRBI*07:01 allele carriers was 8% versus 0.5% in non-carriers. These HLA alleles are present in approximately 15% to 25% of Caucasian, Asian, African, and Hispanic populations and 1% in Japanese populations. Liver function should be monitored in all patients receiving therapy with TYKERB regardless of genotype.
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: In carcinogenicity studies, lapatinib was administered orally for up to 104 weeks at doses of 75 and 150 mg/kg/day in male mice and 75, 150, and 300 mg/kg/day in female mice (approximately 0.7 to 2 times the expected human clinical exposure based on AUC for a clinical dose of 1,250 mg/day plus capecitabine) and 60, 120, 240, and 500 mg/kg/day (approximately 0.6 to 2.3 times the expected human clinical exposure based on AUC) in male rats, and 20, 60, and 180 mg/kg/day (approximately 1.4 to 10 times the expected human clinical exposure based on AUC for a clinical dose of 1,250 mg/day plus capecitabine) in female rats. There was no evidence of carcinogenicity in mice. In male rats, there was an increased incidence of whole body combined hemangiomas and hemangiosarcomas.
Lapatinib was not clastogenic or mutagenic in the Chinese hamster ovary chromosome aberration assay, microbial mutagenesis (Ames) assay, human peripheral lymphocyte chromosome aberration assay or the in vivo rat bone marrow chromosome aberration assay at single doses up to 2,000 mg/kg.
There were no effects on male or female rat mating or fertility at doses up to 120 mg/kg/day in females and 180 mg/kg/day in males (approximately 6.4 times and 2.6 times the expected human clinical exposure based on AUC following 1,250 mg dose of lapatinib plus capecitabine, respectively). The effect of lapatinib on human fertility is unknown. However, when female rats were given oral doses of lapatinib during breeding and through the first 6 days of gestation, a significant decrease in the number of live fetuses was seen at 120 mg/kg/day and in the fetal body weights at greater than or equal to 60 mg/kg/day (approximately 6.4 times and 3.3 times the expected human clinical exposure based on AUC following 1,250 mg dose of lapatinib plus capecitabine, respectively).
Animal Toxicology and/or Pharmacology: In 104-week repeat-dose studies in rodents, severe skin lesions that led to lethality were seen at the highest doses tested (300 mg/kg/day) in male mice and female rats. There was also an increase in renal infarcts and papillary necrosis in female rats at greater than or equal to 60 mg/kg/day and greater than or equal to 180 mg/kg/day, respectively (approximately 7 and 10 times the expected human clinical exposure based on AUC, respectively). The relevance of these findings for humans is uncertain.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image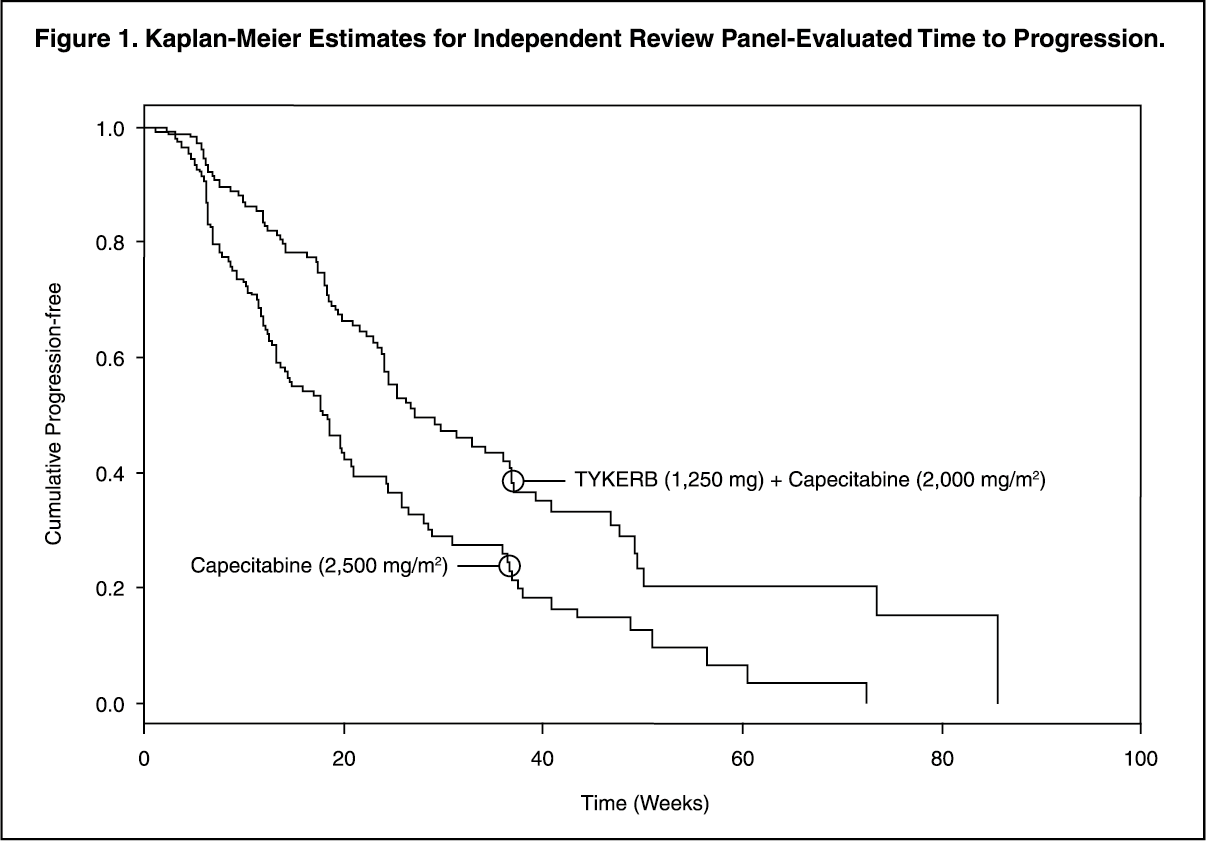 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image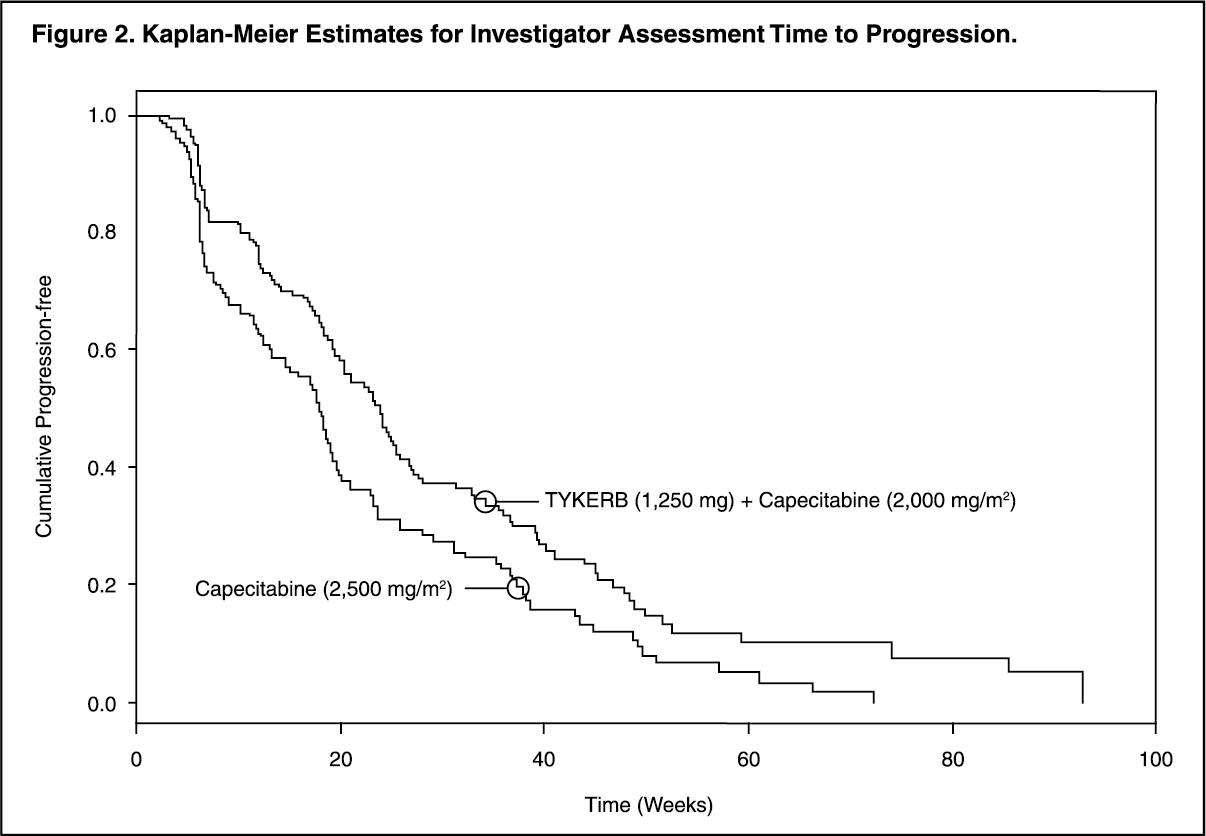 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image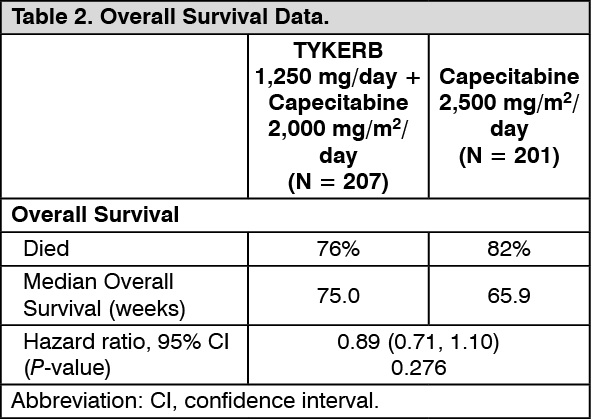 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image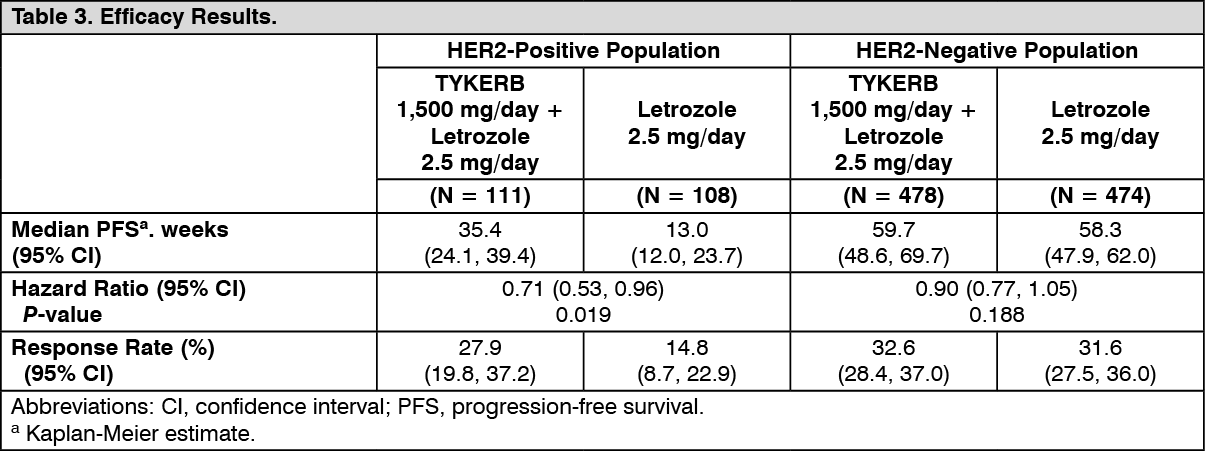 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image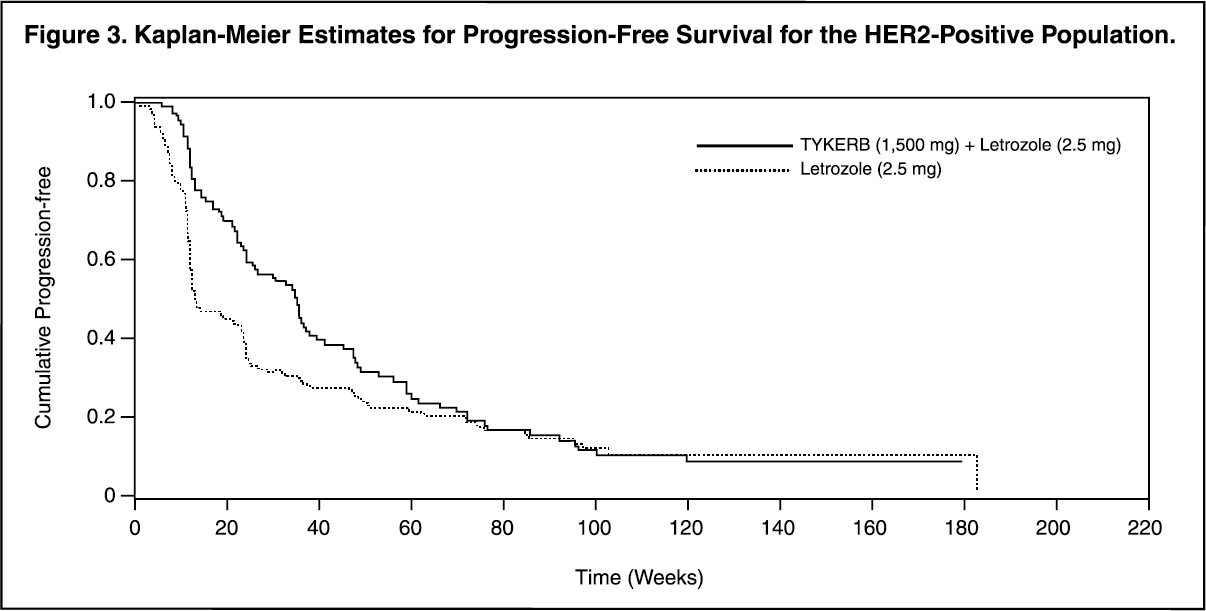 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image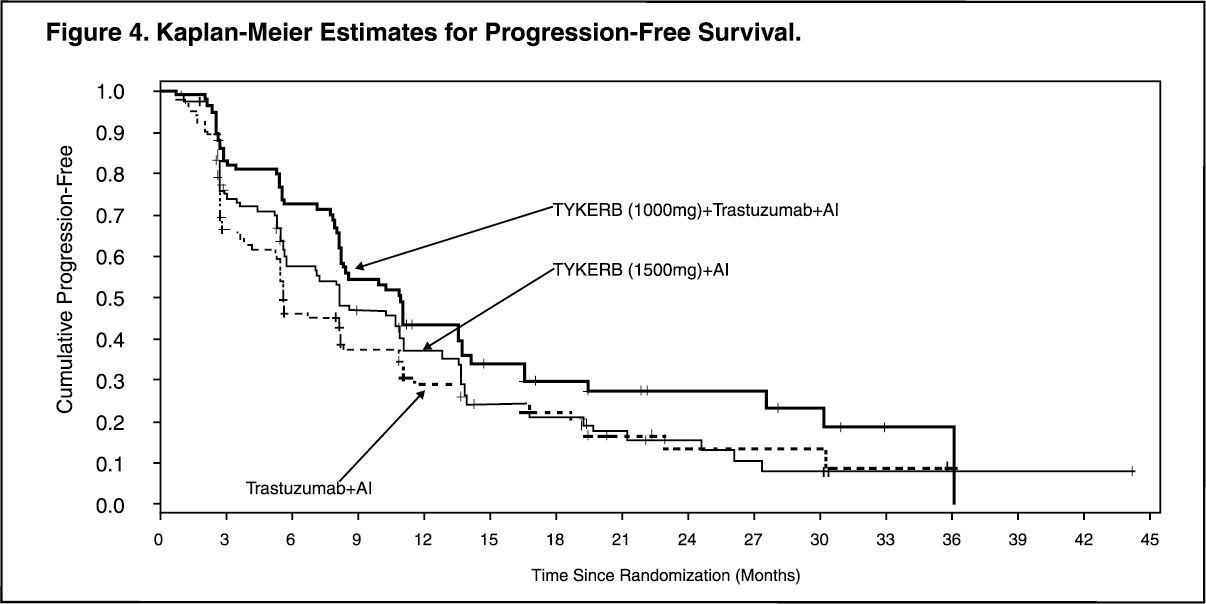 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image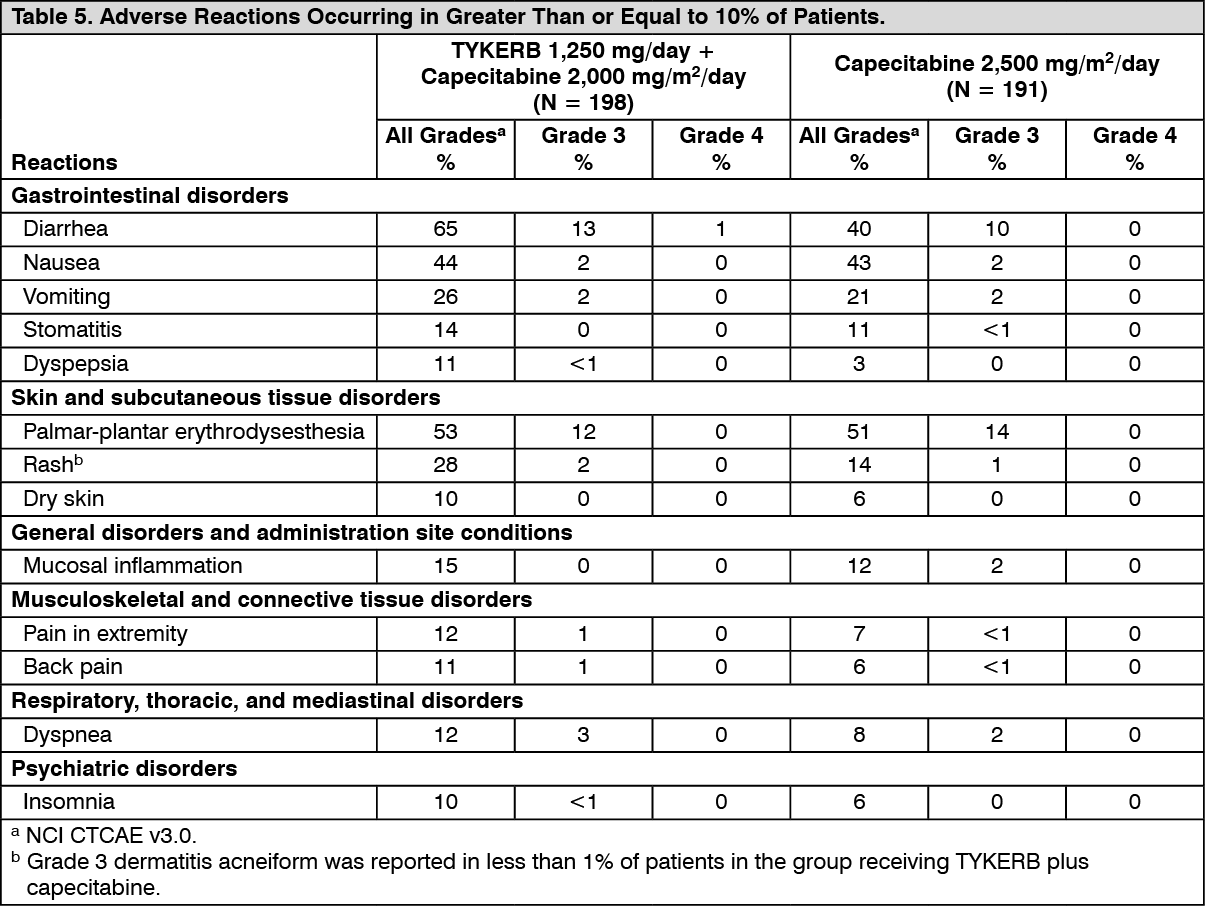 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image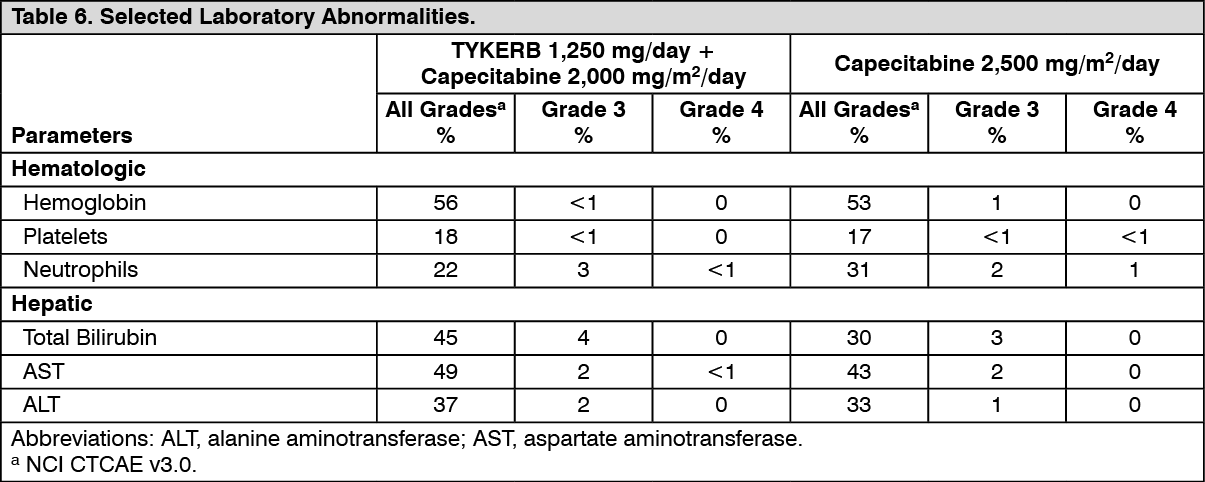 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image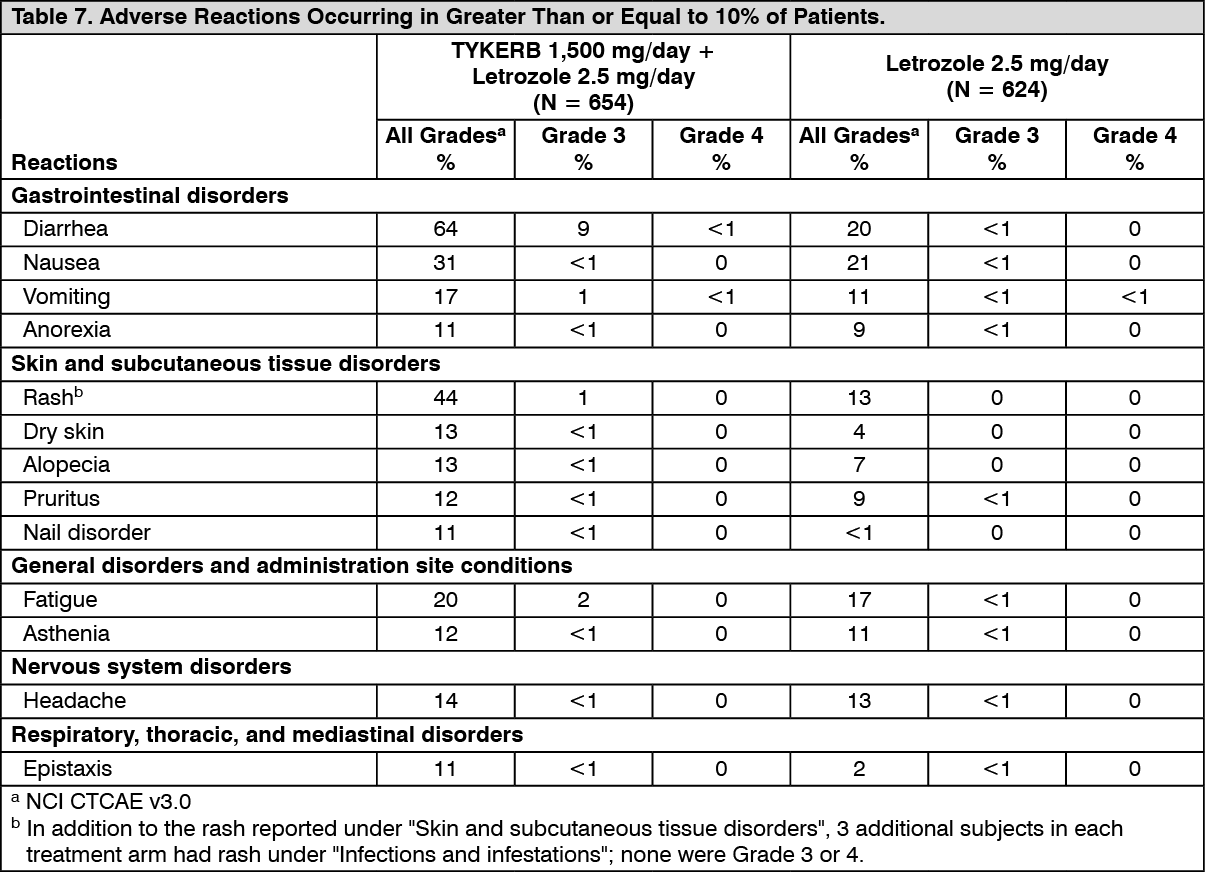 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image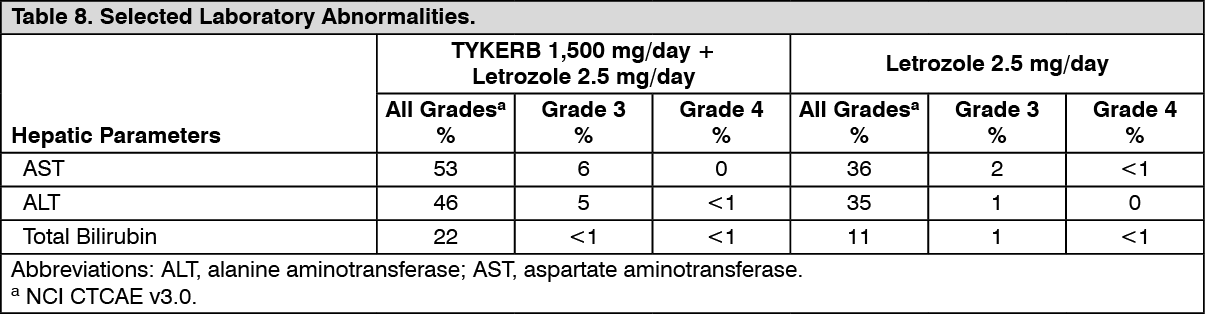 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image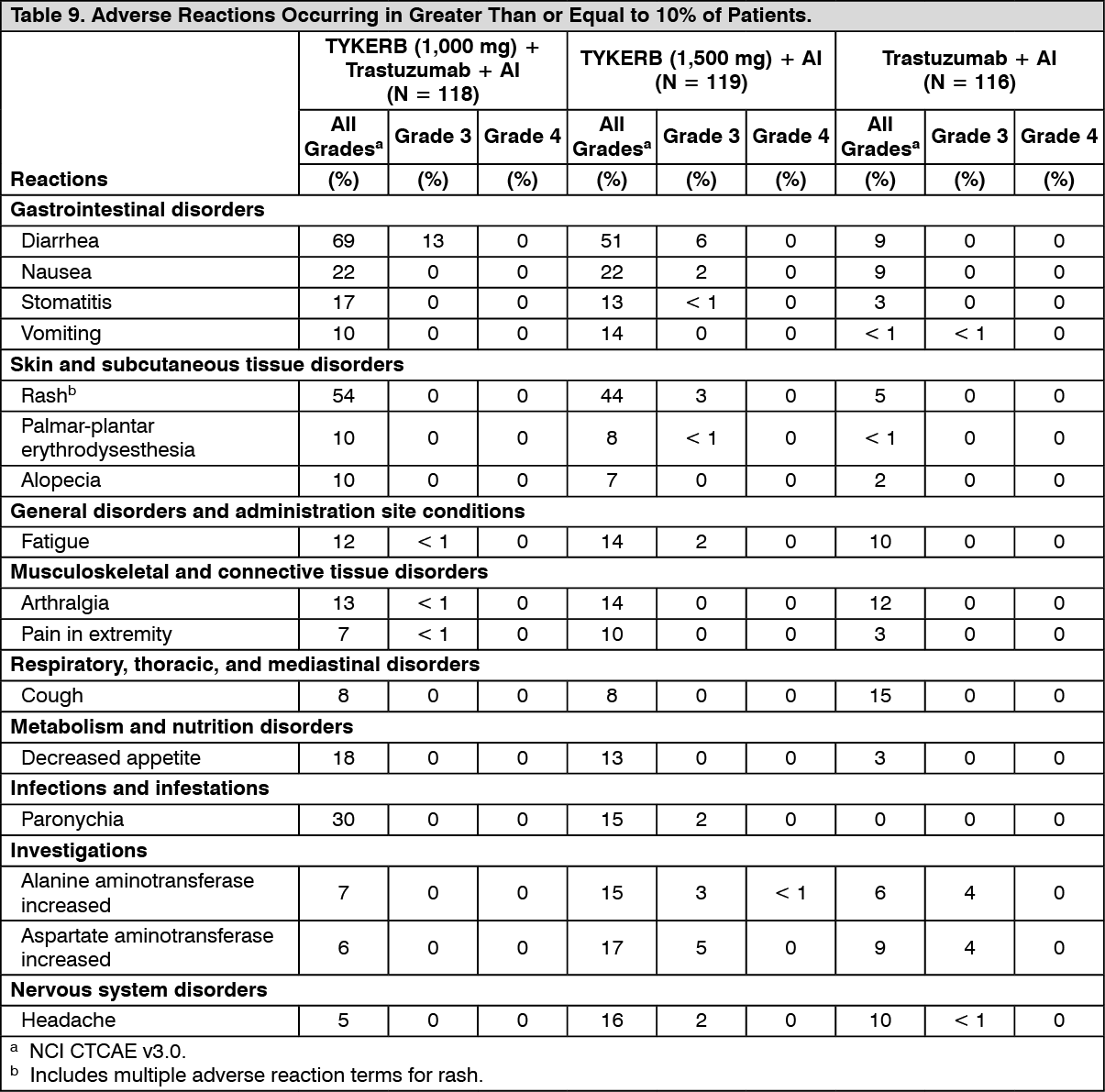 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
