


 Sign Out
Sign Out

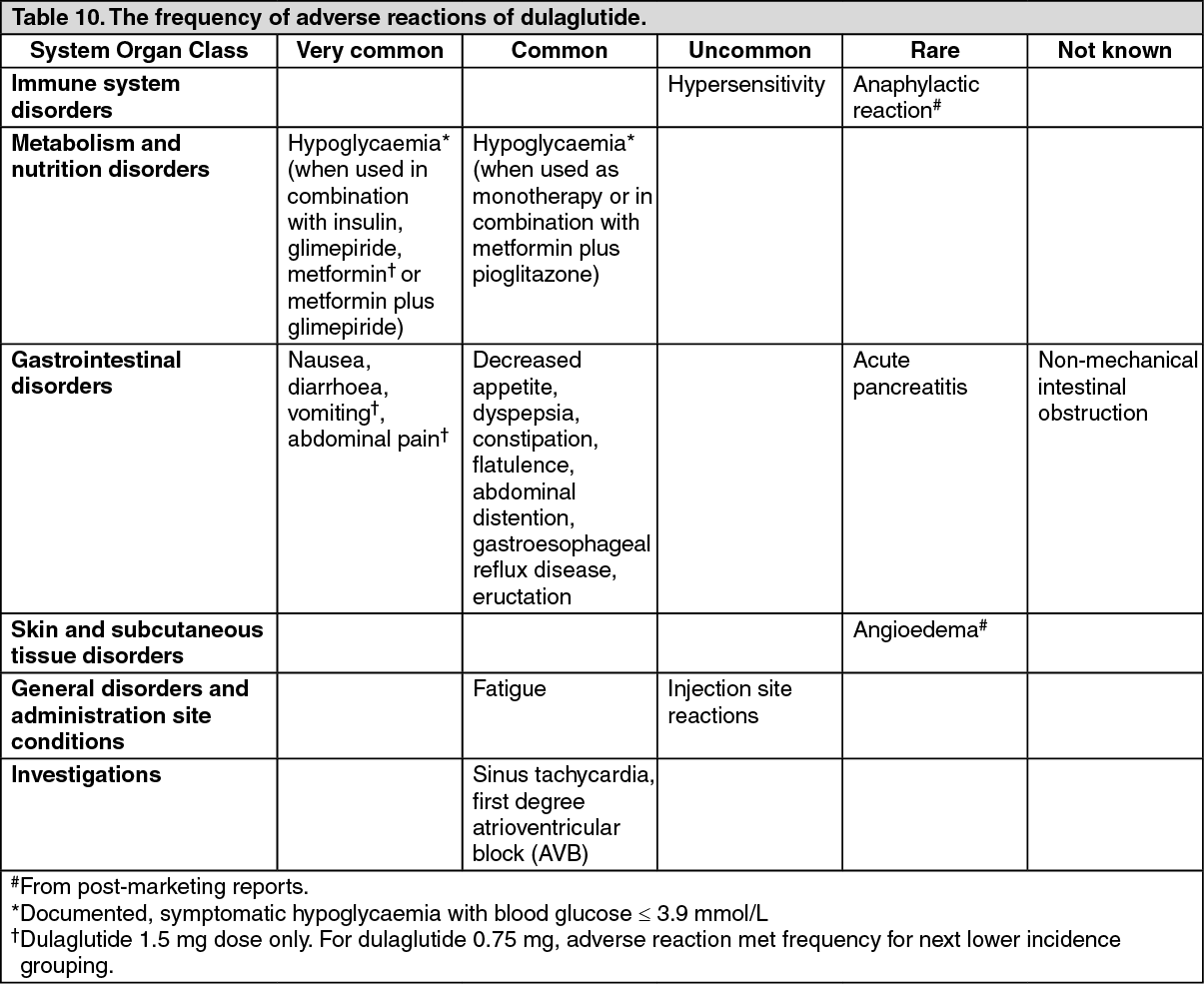
Tabulated list of adverse reactions: The following adverse reactions have been identified based on evaluation of the full duration of the phase II and phase III clinical studies and post-marketing reports and are listed in Table 10 as MedDRA preferred term by system organ class and in order of decreasing incidence (very common: ≥ 1/10; common: ≥ 1/100 to < 1/10; uncommon: ≥ 1/1,000 to < 1/100; rare: ≥ 1/10,000 to < 1/1,000; very rare: < 1/10,000 and not known: cannot be estimated from available data). Within each incidence grouping, adverse reactions are presented in order of decreasing frequency. (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Hypoglycaemia: When dulaglutide 0.75 mg and 1.5 mg were used as monotherapy or in combination with metformin alone or metformin and pioglitazone, the incidences of documented symptomatic hypoglycaemia were 5.9% to 10.9% and the rates were 0.14 to 0.62 events/patient/year, and no episodes of severe hypoglycaemia were reported.
The incidences of documented symptomatic hypoglycaemia when dulaglutide 0.75 mg and 1.5 mg, respectively, were used in combination with a sulphonylurea and metformin were 39.0% and 40.3% and the rates were 1.67 and 1.67 events/patient/year. The severe hypoglycaemia event incidences were 0% and 0.7%, and rates were 0.00 and 0.01 events/patient/year for each dose, respectively. The incidence of documented symptomatic hypoglycaemia when dulaglutide 1.5 mg was used with sulphonylurea alone was 11.3% and the rate was 0.90 events/patient/year, and there were no episodes of severe hypoglycaemia.
The incidence of documented symptomatic hypoglycaemia when dulaglutide 1.5 mg was used in combination with insulin glargine was 35.3% and the rate was 3.38 events/patient/year. The severe hypoglycaemia event incidence was 0.7% and the rate was 0.01 events/patient/year.
The incidences when dulaglutide 0.75 mg and 1.5 mg, respectively, were used in combination with prandial insulin were 85.3% and 80.0% and rates were 35.66 and 31.06 events/patient/year. The severe hypoglycaemia event incidences were 2.4% and 3.4%, and rates were 0.05 and 0.06 events/patient/year.
Gastrointestinal adverse reactions: Cumulative reporting of gastrointestinal events up to 104 weeks with dulaglutide 0.75mg and 1.5 mg, respectively, included nausea (12.9% and 21.2 %), diarrhoea (10.7% and 13.7 %) and vomiting (6.9% and 11.5 %). These were typically mild or moderate in severity and were reported to peak during the first 2 weeks of treatment and rapidly declined over the next 4 weeks, after which the rate remained relatively constant.
In clinical pharmacology studies conducted in patients with type 2 diabetes mellitus up to 6 weeks, the majority of gastrointestinal events were reported during the first 2-3 days after the initial dose and declined with subsequent doses.
Acute pancreatitis: The incidence of acute pancreatitis in Phase II and III clinical studies was 0.07% for dulaglutide compared to 0.14% for placebo and 0.19% for comparators with or without additional background antidiabetic therapy.
Pancreatic enzymes: Dulaglutide is associated with mean increases from baseline in pancreatic enzymes (lipase and/or pancreatic amylase) of 11 % to 21 % (see Precautions). In the absence of other signs and symptoms of acute pancreatitis, elevations in pancreatic enzymes alone are not predictive of acute pancreatitis.
Heart rate increase: Small mean increases in heart rate of 2 to 4 beats per minute (bpm) and a 1.3% and 1.4 % incidence of sinus tachycardia, with a concomitant increase from baseline ≥ 15 bpm, were observed with dulaglutide 0.75mg and 1.5 mg, respectively.
First degree AV block/PR interval prolongation: Small mean increases from baseline in PR interval of 2 to 3 msec and a 1.5% and 2.4 % incidence of first-degree AV block were observed with dulaglutide 0.75 mg and 1.5 mg, respectively.
Immunogenicity: In clinical studies, treatment with dulaglutide was associated with a 1.6 % incidence of treatment emergent dulaglutide anti-drug antibodies, indicating that the structural modifications in the GLP-1 and modified IgG4 parts of the dulaglutide molecule, together with high homology with native GLP-1 and native IgG4, minimise the risk of immune response against dulaglutide. Patients with dulaglutide anti-drug antibodies generally had low titres, and although the number of patients developing dulaglutide anti-drug antibodies was low, examination of the phase III data revealed no clear impact of dulaglutide anti-drug antibodies on changes in HbA1c. None of the patients with systemic hypersensitivity developed dulaglutide anti-drug antibodies.
Hypersensitivity: In the phase II and phase III clinical studies, systemic hypersensitivity events (e.g., urticaria, edema) were reported in 0.5 % of patients receiving dulaglutide. Cases of anaphylactic reaction have been rarely reported with marketed use of dulaglutide.
Injection site reactions: Injection site adverse events were reported in 1.9 % of patients receiving dulaglutide. Potentially immune-mediated injection site adverse events (e.g., rash, erythema) were reported in 0.7 % of patients and were usually mild.
Discontinuation due to an adverse event: In studies of 26 weeks duration, the incidence of discontinuation due to adverse events was 2.6% (0.75 mg) and 6.1% (1.5 mg) for dulaglutide versus 3.7 % for placebo. Through the full study duration (up to 104 weeks), the incidence of discontinuation due to adverse events was 5.1% (0.75 mg) and 8.4 % (1.5 mg) for dulaglutide. The most frequent adverse reactions leading to discontinuation for 0.75 mg and 1.5 mg dulaglutide, respectively, were nausea (1.0%, 1.9 %), diarrhoea (0.5%, 0.6 %), and vomiting (0.4%, 0.6 %), and were generally reported within the first 4-6 weeks.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions.
View ADR Monitoring Form