Pharmacotherapeutic group: Immunosuppressants, interleukin inhibitors.
ATC code: L04AC16.
Pharmacology: Pharmacodynamics: Mechanism of action: Guselkumab is a human IgG1λ monoclonal antibody (mAb) that binds selectively to the interleukin 23 (IL-23) protein with high specificity and affinity. IL-23, a regulatory cytokine, affects the differentiation, expansion, and survival of T cell subsets, (e.g., Th17 cells and Tc17 cells) and innate immune cell subsets, which represent sources of effector cytokines, including IL-17A, IL-17F and IL-22 that drive inflammatory disease. In humans, selective blockade of IL-23 was shown to normalize production of these cytokines.
Levels of IL-23 are elevated in the skin of patients with plaque psoriasis. In
in vitro models, guselkumab was shown to inhibit the bioactivity of IL-23 by blocking its interaction with cell surface IL-23 receptor, disrupting IL-23-mediated signaling, activation and cytokine cascades. Guselkumab exerts clinical effects in plaque psoriasis through blockade of the IL-23 cytokine pathway.
Pharmacodynamic effects: In a Phase 1 study, treatment with guselkumab resulted in reduced expression of IL-23/Th17 pathway genes and psoriasis-associated gene expression profiles, as shown by analyses of mRNA obtained from lesional skin biopsies of psoriatic subjects at Week 12 compared to baseline. In the same Phase 1 study, treatment with guselkumab resulted in improvement of histological measures of psoriasis at Week 12, including reductions in epidermal thickness and T-cell density. In addition, reduced serum IL-17A, IL-17F and IL-22 levels compared to placebo were observed in guselkumab-treated subjects in Phase 2 and Phase 3 studies in plaque psoriasis. These results are consistent with the clinical benefit observed with guselkumab treatment in plaque psoriasis.
In Phase 3 studies in psoriatic arthritis, evaluated subjects had elevated serum levels of acute phase proteins C-reactive protein, serum amyloid A and IL-6, and Th17 effector cytokines IL-17A, IL-17F and IL-22 at baseline. Guselkumab decreased levels of these proteins within 4 weeks of initiation of treatment. By Week 24, guselkumab further reduced the levels of these proteins compared to baseline and also to placebo. In guselkumab-treated subjects, serum IL-17A and IL-17F levels were similar to those observed in a demographically matched healthy cohort at Week 24.
Immunogenicity: As with all therapeutic proteins, there is the potential for immunogenicity. The immunogenicity of TREMFYA was evaluated using a sensitive and drug-tolerant immunoassay.
Plaque psoriasis: In pooled Phase 2 (PSO2001) and Phase 3 (VOYAGE 1, VOYAGE 2 and NAVIGATE) analyses, fewer than 6% of subjects treated with TREMFYA developed antidrug antibodies in up to 52 weeks of treatment. Of the subjects who developed antidrug antibodies, approximately 7% had antibodies that were classified as neutralizing which equates to 0.4% of all subjects treated with TREMFYA. In pooled Phase 3 analyses, approximately 15% of patients treated with TREMFYA developed antidrug antibodies in up to 264 weeks of treatment. Of the subjects who developed antidrug antibodies, approximately 5% had antibodies that were classified as neutralizing which equates to 0.76% of all subjects treated with TREMFYA. Antidrug antibodies were not associated with lower efficacy or development of injection-site reactions.
Psoriatic arthritis: In pooled Phase 3 (DISCOVER 1 and DISCOVER 2) analyses up to Week 52, 4.5% (n=49) of subjects treated with TREMFYA developed antidrug antibodies. Of these subjects, 5 had antibodies that were classified as neutralizing antibodies, and 3 developed injection site reactions through Week 52. Overall, the small number of subjects who were positive for antibodies to guselkumab limits definitive conclusion of the effect of immunogenicity on the pharmacokinetics and efficacy of guselkumab.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to TREMFYA with the incidences of antibodies to other products may be misleading.
Clinical studies: Clinical efficacy-plaque psoriasis (Adults): The efficacy and safety of TREMFYA was assessed in three Phase 3, multicenter, randomized, double-blind, placebo and/or active controlled studies (VOYAGE 1, VOYAGE 2 and NAVIGATE and ORION) in adult subjects with moderate to severe chronic plaque-type psoriasis eligible for systemic or phototherapy.
The studies enrolled adult subjects (≥ 18 years) with moderate to severe plaque psoriasis (with or without PsA) defined by Investigator's Global Assessment (IGA) ≥ 3, a Body Surface Area (BSA) involvement ≥ 10%, and Psoriasis Area and Severity Index (PASI) score ≥ 12, and were candidates for either systemic therapy or phototherapy for psoriasis. No concomitant antipsoriatic therapies for psoriasis were allowed during the study. Subjects with guttate, erythrodermic, or pustular psoriasis were excluded from the studies. The efficacy of TREMFYA was evaluated with respect to overall skin disease, regional disease (scalp, hand and foot, and nails) and patient reported outcomes (PROs).
The IGA is a 5-category scale: 0 = cleared, 1 = minimal, 2 = mild, 3 = moderate, 4 = severe, that indicates the physician's overall assessment of psoriasis focusing on plaque thickness/induration, erythema and scaling.
The PASI is a composite score that assesses the fraction of body surface area involved with psoriasis and the severity of psoriatic lesions within the affected regions (plaque thickness/induration, erythema, and scaling). PASI numeric scores range from 0 to 72, with higher scores representing more severe disease.
Other key efficacy assessments included: The Scalp-specific Investigator Global Assessment (ss-IGA), is used to evaluate the disease severity of scalp psoriasis. The lesions are assessed based on a 5 point scale in terms of clinical signs of redness, thickness, and scaliness with 0 indicating absence of disease and a score of 4 representing severe disease.
Physician's Global Assessment of Hands and/or Feet (hf-PGA), assesses the severity of hand and foot psoriasis. The plaques are scored on a 5-point scale with a score of 0 indicating clear to 4 being severe.
The Nail Psoriasis Severity Index (NAPSI), a physician-assessed score that measures the severity of fingernail involvement. The scale consists of 4 components of nail matrix disease and 4 components of nail bed disease with scores from 0 to 8, with a lower score representing milder disease. Fingernail Physician's Global Assessment (f-PGA), is also a physician assessed score that is used to evaluate fingernail psoriasis on a scale of 0 to 4 with 4 indicating severe disease.
The Psoriasis Symptoms and Signs Diary (PSSD), includes patient reported outcomes that were designed to measure the severity of psoriasis symptoms (itch, pain, burning, skin tightness, stinging) and signs (skin dryness, cracking, shedding or flaking, scaling, redness and bleeding) using 0 to 10 numerical rating scale for the assessment of treatment benefit. Symptom summary score and sign summary score were derived, ranging from 0 to 100. A higher score represented more severe disease.
The Dermatology Life Quality Index (DLQI), a dermatology-specific quality of life instrument designed to assess the impact of the disease on a patient's quality of life. DLQI scores range from 0 to 30, with a lower score representing a better quality of life.
The SF-36, a health survey questionnaire consisting of multi-item scales measuring 8 health concepts. The SF-36 yields composite scores that provide a measure of disease impact on physical and mental health status. Higher SF-36 scores indicate a better quality of life.
The Hospital Anxiety and Depression Scale (HADS), a self-rating tool developed to evaluate psychological measures in patients with physical ailments. It consists of 2 subscales, one measuring anxiety (A-scale) and one measuring Depression (D-scale), which are scored separately. Lower HADS scores correspond to lesser psychological impairment.
The Work Limitations Questionnaire (WLQ), a 25-item, self-administered questionnaire that was used to measure the impact of chronic health conditions on job performance and work productivity among employed populations. The WLQ assesses four aspects of work and productivity: Physical Demands, Time Management, Mental-Interpersonal Demand, and Output Demand. The four subscales range from 0-100 with the lower score indicating fewer work limitations.
Placebo- and adalimumab-controlled studies - VOYAGE 1 and VOYAGE 2: VOYAGE 1 evaluated the safety and efficacy of TREMFYA vs. placebo and adalimumab in 837 subjects with plaque psoriasis. Subjects randomized to TREMFYA received TREMFYA 100 mg at Weeks 0 and 4 and every 8 weeks thereafter. Subjects randomized to adalimumab received adalimumab 80 mg at Week 0 and 40 mg at Week 1 subcutaneously followed by 40 mg every other week thereafter through Week 47. All subjects, including those randomized to adalimumab at Week 0, received TREMFYA 100 mg at Week 52 and every 8 weeks thereafter. Subjects randomized to placebo received TREMFYA at Weeks 16, 20 and every 8 weeks thereafter.
VOYAGE 2 evaluated the safety and efficacy of TREMFYA vs. placebo and adalimumab in 992 subjects with plaque psoriasis. Subjects randomized to TREMFYA received TREMFYA 100 mg at Weeks 0, 4, 12 and 20. Subjects randomized to adalimumab received adalimumab 80 mg at Week 0 and 40 mg at Week 1 subcutaneously followed by 40 mg every other week thereafter through Week 23. Subjects randomized to placebo received TREMFYA 100 mg at Weeks 16 and 20. To evaluate the therapeutic benefit of maintenance dosing with TREMFYA, subjects randomized to TREMFYA at Week 0 who were PASI 90 responders at Week 28 were re-randomized to either continue treatment with TREMFYA maintenance therapy or withdrawal of therapy. Withdrawal subjects re-initiated TREMFYA (dosed at time of retreatment, 4 weeks later and every 8 weeks thereafter) when they experienced at least a 50% loss of their week 28 PASI improvement. Subjects randomized to adalimumab at Week 0 who were PASI 90 non-responders received TREMFYA at Weeks 28, 32 and every 8 weeks thereafter. All subjects started to receive open-label TREMFYA every 8 weeks at Week 76.
The co-primary endpoints in VOYAGE 1 and VOYAGE 2 were the proportions of subjects who achieved an IGA score of cleared (0) or minimal (1) and the proportions of subjects who achieved a PASI 90 response at Week 16, comparing the TREMFYA group and the placebo group. For both studies, secondary endpoints comparing TREMFYA and adalimumab groups included the proportions of subjects who achieved an IGA score of cleared (0) or minimal (1), a PASI 90 and a PASI 75 response at Week 16; and the proportions of subjects achieving an IGA score of cleared (0), an IGA score of cleared or minimal (0 or 1), PASI 75, PASI 90 and a PASI 100 response at Week 24, and at Week 48 for VOYAGE 1.
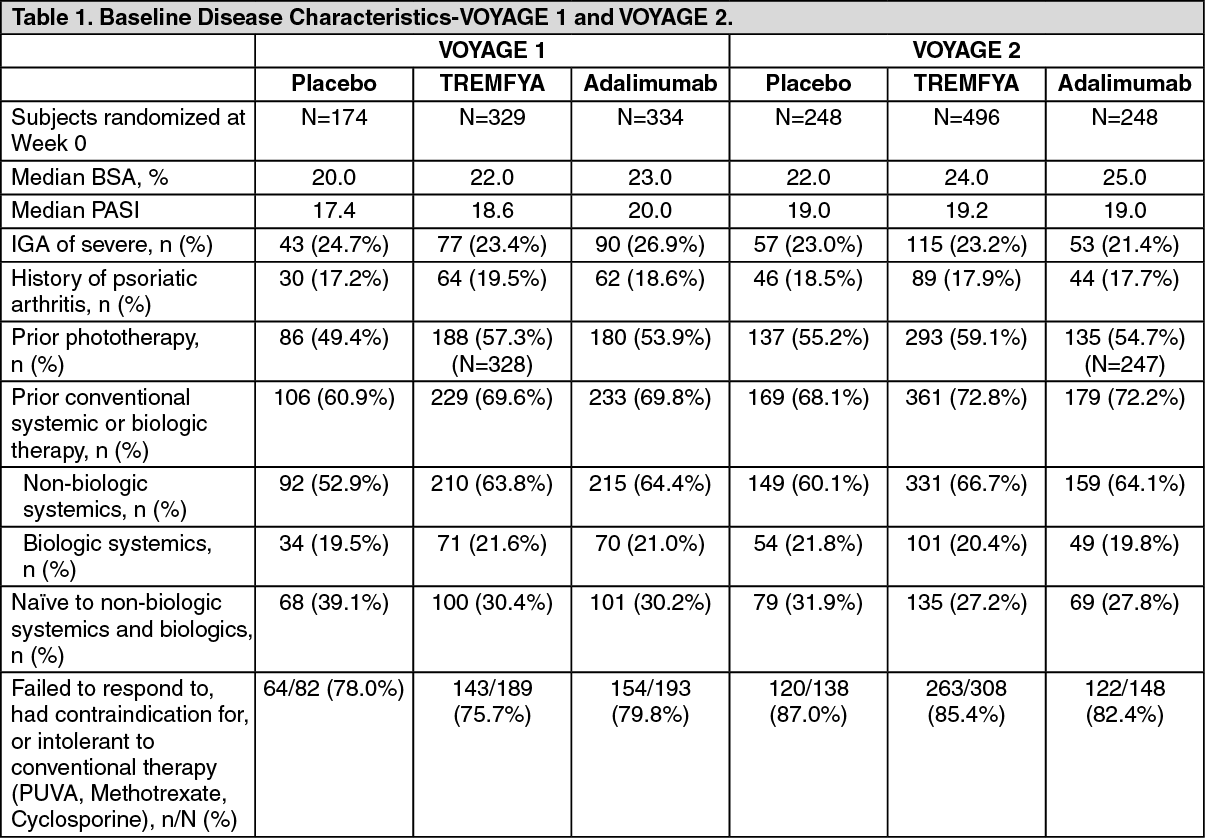
Baseline disease characteristics were generally consistent across all treatment groups in VOYAGE 1 and VOYAGE 2 (see Table 1). The majority of subjects were male and white. The mean age was approximately 44 years, and mean weight was approximately 90 kg. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Baseline disease characteristics were consistent for the study populations in VOYAGE 1 and 2 with a median BSA of 22% and 24%, a median baseline PASI score of 19 for both studies, a baseline IGA score of severe for 25% and 23% of subjects, and a history of psoriatic arthritis for 19% and 18% of subjects, respectively.
Of all subjects who were included in the VOYAGE 1 and 2 studies, 32% and 29% were naive to conventional systemic and biologic systemic therapy; 54% and 57% had received prior phototherapy, and 62% and 64% had received prior conventional systemic therapy, respectively. In both studies, 21% had received prior biologic systemic therapy, including 11% who had received at least one anti-tumor necrosis factor alpha (TNFα) agent, and approximately 10% who had received an anti-IL-12/IL-23 agent.
Summary of clinical outcomes: PASI and IGA outcomes, VOYAGE 1 and VOYAGE 2: In both the VOYAGE 1 and VOYAGE 2 studies, a significantly greater proportion of subjects randomized to treatment with TREMFYA achieved a PASI 90 response and IGA cleared or minimal (0 or 1) response versus placebo at Week 16 (p < 0.001 for all comparisons) (see Table 2).
TREMFYA demonstrated superiority to adalimumab as evaluated by efficacy endpoints of PASI 75, PASI 90 and IGA cleared or minimal (0 or 1) at Week 16 in both studies (p < 0.001 for all comparisons). TREMFYA also demonstrated superiority to adalimumab on PASI 75, PASI 90, PASI 100, IGA cleared (0), and IGA cleared or minimal (0 or 1) at Week 24 in both studies and at Week 48 in VOYAGE 1 (p < 0.001 for all comparisons) (see Table 2).
Response rates to TREMFYA were similar among the subgroups defined by age, gender, race, body weight, plaques location and baseline PASI score. Response rates in subjects with concurrent psoriatic arthritis at baseline were similar to those in the overall plaque psoriasis population. TREMFYA was efficacious in systemic treatment-naive, systemic treatment-exposed, biologic-naive, and biologic-exposed subjects. (See Table 2.)
Click on icon to see table/diagram/image
Response over time: TREMFYA demonstrated rapid onset of efficacy, with a significantly higher percent improvement in PASI as compared with placebo as early as Week 2 (p < 0.001). The percentage of subjects achieving a PASI 90 response was numerically higher for TREMFYA than adalimumab starting at Week 8 with the difference reaching a maximum around Week 20 (VOYAGE 1 and VOYAGE 2) and maintained through Week 48 (VOYAGE 1). In VOYAGE 1, for subjects receiving continuous TREMFYA treatment, PASI 90 response was maintained from Week 52 to Week 252. (See Figures 1 and 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Maintenance and durability of response: To evaluate the maintenance and durability of response, subjects originally randomized to TREMFYA and who were PASI 90 responders at Week 28 in the VOYAGE 2 study were re-randomized to continue maintenance treatment with TREMFYA or be withdrawn from therapy (i.e. placebo). At Week 48, 88.6% of subjects in the continuous maintenance treatment group were PASI 90 responders compared with 36.8% in the withdrawal group (p < 0.001). Loss of PASI 90 response was noted as early as 4 weeks after withdrawal of therapy with the median time to loss of PASI 90 of approximately 15 weeks.
Therefore, a maintenance regimen of every 8 weeks is recommended.
Efficacy of retreatment: In VOYAGE 2, among subjects who were withdrawn from treatment and subsequently re-initiated TREMFYA, 80% regained a PASI 90 response when assessed 20 weeks after initiation of retreatment.
Efficacy and safety in patients switching from adalimumab to TREMFYA: In VOYAGE 2, among 112 adalimumab subjects who failed to achieve a PASI 90 response at Week 28, 66% and 76% achieved a PASI 90 response after 20 and 44 weeks of treatment with TREMFYA, respectively.
No new safety findings were observed in patients who switched from adalimumab to guselkumab.
Analyses related to regional psoriasis disease: Significant improvements were seen in psoriasis involving the scalp, hands and feet, and nails in subjects randomized to TREMFYA compared to placebo at Week 16. TREMFYA demonstrated superiority compared to adalimumab for treatment of psoriasis involving the scalp, or hands and feet at Week 24 (VOYAGE 1 and VOYAGE 2) and Week 48 (VOYAGE 1) (p ≤ 0.001 for all comparisons, except p < 0.05 for hf-PGA 0/1 and f-PGA 0/1 at Week 48 in VOYAGE 1, and for hf-PGA 0/1 at Week 24 in VOYAGE 2). (See Table 3.)
Click on icon to see table/diagram/image
Scalp psoriasis: At Week 16, in subjects with a baseline ss-IGA score ≥ 2, 83.4% and 80.6% in the TREMFYA group in VOYAGE 1 and VOYAGE 2, respectively, achieved a ss-IGA score of 0/1 and at least a 2-grade improvement from baseline compared to 14.5% and 10.9% in the placebo group, respectively (p < 0.001 for all comparisons).
Additionally, at Week 24, 84.5% and 85.3% in the TREMFYA group in VOYAGE 1 and VOYAGE 2, respectively, achieved a ss-IGA score of 0/1 and at least a 2-grade improvement from baseline compared to 69.2% and 67.5% in the adalimumab group, respectively (p < 0.001 for all comparisons). At Week 48, this outcome was achieved in 78.3% of TREMFYA subjects compared to 60.5% of adalimumab subjects in VOYAGE 1 (p < 0.001).
Hand/foot psoriasis: At Week 16, in subjects with a baseline hf-PGA score ≥ 2, 73.3% and 77.2% in the TREMFYA group in VOYAGE 1 and VOYAGE 2, respectively, achieved a hf-PGA score of 0/1 and at least a 2-grade improvement from baseline compared to 14.0% and 14.3% in the placebo group, respectively (p < 0.001 for all comparisons).
Additionally, at Week 24, 78.9% and 81.6% in the TREMFYA group in VOYAGE 1 and VOYAGE 2, respectively, achieved a hf-PGA score of 0/1 and at least a 2-grade improvement from baseline compared with 56.8% and 66.1% in the adalimumab group, respectively (p = 0.001 for VOYAGE 1; p < 0.05 for VOYAGE 2). At Week 48, this outcome was achieved in 75.6% of TREMFYA subjects compared to 62.1% of adalimumab subjects in VOYAGE 1 (p < 0.05).
Nail psoriasis: At Week 16, in subjects with a baseline f-PGA score ≥ 2, 39.1% and 52.0% in the TREMFYA group in VOYAGE 1 and VOYAGE 2, respectively, achieved a f-PGA score of 0/1 compared to 15.9% and 14.6% in the placebo group, respectively (p < 0.001 for all comparisons). In subjects with a baseline NAPSI score > 0, the median percent improvement in NAPSI is 33.3 and 50.0 in VOYAGE 1 and VOYAGE 2, respectively, for the TREMFYA group and 0 for both studies for the placebo group (p < 0.001 for all comparisons).
At Week 48, a significantly higher proportion of subjects (74.7%) treated with TREMFYA achieved a f-PGA score of 0/1 compared to subjects (61.8%) randomized to adalimumab in VOYAGE 1 (p < 0.05).
No significant differences were seen in f-PGA at Week 24 and in NAPSI at Weeks 24 and 48 in subjects randomized to TREMFYA compared to adalimumab.
Patient reported outcomes: In the VOYAGE 1 and VOYAGE 2 studies, patient reported outcomes of psoriasis symptoms and signs were assessed with the PSSD, and disease specific health related quality of life was evaluated with the DLQI at Weeks 16, 24, 48, 76, 100, 124 and 156. In addition, the VOYAGE 2 study also included assessments of general health status with the SF-36, anxiety and depression with the HADS and work limitations with the WLQ in subjects treated with TREMFYA.
Psoriasis Symptoms and Signs Diary (PSSD): In VOYAGE 1 and VOYAGE 2, TREMFYA-treated subjects demonstrated significantly greater improvement in both PSSD symptom and signs scores from baseline compared to placebo at Week 16 and compared to adalimumab at Week 24 (VOYAGE 1 and VOYAGE 2) and Week 48 (VOYAGE 1) (see Table 4). TREMFYA demonstrated greater improvement as compared to placebo as early as Week 2.
A significantly greater proportion of subjects treated with TREMFYA achieved a clinically meaningful improvement (≥ 40 points reduction) from baseline in PSSD symptom score and signs score compared to placebo at Week 16, and compared to adalimumab at Week 24 (VOYAGE 1 and VOYAGE 2) and Week 48 (VOYAGE 1) (p ≤ 0.002, for all comparisons). A significantly greater proportion of subjects treated with TREMFYA achieved PSSD symptom and signs score of 0 (symptom free and sign free) compared to placebo at Week 16, and compared to adalimumab at Week 24 (VOYAGE 1 and VOYAGE 2) and Week 48 (VOYAGE 1) (p < 0.001, for all comparisons, except p = 0.003 for signs score of 0 at Week 24 in VOYAGE 2) (see Table 4).
Significantly greater improvements in each of the individual items within the PSSD symptom scale (itching, pain, burning, stinging and skin tightness) and PSSD sign scale (skin dryness, cracking, scaling, shedding or flaking, redness and bleeding) were demonstrated in TREMFYA-treated subjects when compared to placebo at Week 16, and when compared to adalimumab at Week 24 (VOYAGE 1 and VOYAGE 2) and Week 48 (VOYAGE 1).
In VOYAGE 1, for subjects receiving continuous TREMFYA treatment, improvements in PSSD scores were maintained from Week 52 through Week 252.
Dermatology Life Quality Index: Significantly greater improvements in the DLQI from baseline were observed in subjects treated with TREMFYA compared to placebo at Week 16 (for all comparisons, p < 0.001). A significantly greater proportion of subjects treated with TREMFYA achieved a DLQI 0 or 1 (no impact of psoriasis on health-related quality of life) compared to placebo at Week 16, and compared to adalimumab at Week 24 (VOYAGE 1 and VOYAGE 2) and Week 48 (VOYAGE 1) (for all comparisons, p < 0.001) (see Table 4).
In VOYAGE 1, for subjects receiving continuous TREMFYA treatment, improvements in DLQ1 scores were maintained from Week 52 through Week 252. (See Table 4.)
Click on icon to see table/diagram/image
SF-36: At Week 16, subjects treated with TREMFYA in VOYAGE 2 showed greater improvement from baseline in the SF-36 physical and mental component summary score compared to subjects treated with placebo (p < 0.001). The improvement in SF-36 physical and mental component summary score was maintained through Week 252 among subjects randomized to TREMFYA maintenance therapy.
Hospital Anxiety and Depression Scale (HADS): Both anxiety and depression scores were significantly reduced in subjects treated with TREMFYA at Week 16 in VOYAGE 2 compared with subjects randomized to placebo (p < 0.001). HADS improvements were maintained through Week 252 among subjects randomized to TREMFYA maintenance therapy. (See Table 5.)
Click on icon to see table/diagram/image
Work Limitations Questionnaire: The WLQ in VOYAGE 2 showed that work productivity improved significantly more in subjects randomized to TREMFYA at Week 16 compared with subjects randomized to placebo as measured by the four WLQ subscales (Physical Demands, Time Management, Mental-Interpersonal, and Output Demands). The improvements in WLQ were maintained through Week 252 among subjects randomized to maintenance therapy. (See Table 6.)
Click on icon to see table/diagram/image
Active-controlled study in ustekinumab inadequate responder-NAVIGATE: NAVIGATE evaluated the efficacy and safety of switching to TREMFYA in 268 subjects who had not achieved an adequate response (defined as IGA ≥ 2) to ustekinumab at Week 16 after initial treatment with ustekinumab (dosed at Week 0 and Week 4). Subjects were randomized to either continue ustekinumab treatment every 12 weeks or to begin TREMFYA 100 mg at Weeks 16, 20, and every 8 weeks thereafter. The primary endpoint was the number of post-randomization visits between Weeks 12 and 24 at which subjects achieved an IGA of cleared or minimal (0 or 1) and had at least a 2-grade improvement. Secondary endpoints included the number of post-randomization visits between Weeks 12 and 24 at which subjects achieved a PASI 90 response, the number of post-randomization visits between Weeks 12 and 24 at which subjects achieved an IGA of 0 and the proportion of subjects who achieved an IGA of cleared or minimal (0 or 1) and at least a 2-grade improvement at 12 weeks post-randomization. Baseline characteristics for randomized subjects were similar to those observed in VOYAGE 1 and VOYAGE 2.
In subjects with an inadequate response to ustekinumab, significantly greater improvement of efficacy was observed in subjects who switched to TREMFYA treatment compared to subjects who continued ustekinumab treatment. Between 12 and 24 weeks after randomization, TREMFYA-treated subjects achieved an IGA score of clear or minimal (0 or 1) with at least a 2-grade improvement twice as often as ustekinumab-treated subjects (mean 1.5 vs 0.7 visits at which this outcome was observed respectively, p < 0.001). Similar outcomes were observed for the number of visits at which subjects achieved a PASI 90 response or an IGA score of cleared (0). At 12 weeks post-randomization, greater proportions of subjects in the TREMFYA group compared to the ustekinumab group also achieved an IGA score of cleared or minimal (0 or 1) and at least a 2-grade improvement (31.1% vs. 14.3%, respectively; p = 0.001) and a PASI 90 response (48% vs 23%, respectively; p <0.001). Differences in response rates between TREMFYA and ustekinumab-treated subjects were noted as early as 4 weeks after randomization and reached a maximum 24 weeks after randomization (see Figure 3).
Click on icon to see table/diagram/image
No new safety findings were observed in patients who switched from ustekinumab to TREMFYA. (See Figure 4.)
Click on icon to see table/diagram/image
Placebo-controlled study with Pre-filled pen-ORION: ORION evaluated the efficacy, safety, PK, immunogenicity, usability, and acceptability of TREMFYA delivered with a pre-filled pen. In this study, 78 subjects were randomized to receive either TREMFYA (100 mg at Weeks 0 and 4 and every 8 weeks thereafter), or placebo. Baseline characteristics for randomized subjects were comparable to those observed in VOYAGE 1 and VOYAGE 2. The co-primary endpoints were the proportion of subjects who achieved an IGA score of 0 or 1 at Week 16 and the proportion of subjects who achieved a PASI 90 response at Week 16. The secondary endpoints included the proportion of subjects who achieved an IGA score 0 at Week 16 and the proportion of subjects who achieved a PASI 100 response at Week 16.
A significantly greater proportion of subjects in the TREMFYA group achieved an IGA score of 0 or 1 or a PASI 90 response at Week 16 (80.6% and 75.8%, respectively, p < 0.001 for both endpoints) than in the placebo group (0% for both endpoints). The proportion of subjects who achieved an IGA score of 0 at Week 16 was significantly higher in the TREMFYA group compared to the placebo group (56.5% vs. 0%; p < 0.001). The proportion of subjects who achieved a PASI 100 response at Week 16 was significantly higher in the TREMFYA group compared to the placebo group (50.0% vs. 0%; p < 0.001).
Patient Experience: Subject experience with the pre-filled pen was assessed on a scale of 0 (worst) to 10 (best) using a validated Self-Injection Assessment Questionnaire (SIAQ) based on subject responses across 6 domains (feelings about injections, self-image, self-confidence, pain and skin reactions during or after the injection, ease of use of the self-injection device, and satisfaction with self-injection) at weeks 0, 4 and 12. At week 12, the mean score for "Satisfaction with Self Injection" was 9.18 (with 10 indicating "Very Satisfied") and the mean score for "Ease of Use" was 9.24 (with 10 indicating "Very Easy"). The mean scores for the other domains at week 12 ranged from 8.43 to 9.84.
Active-controlled study with secukinumab - ECLIPSE: The efficacy and safety of TREMFYA were also investigated in a double-blind study compared to secukinumab. Patients were randomized to receive TREMFYA (N=534; 100 mg at Week 0, 4 and every 8 weeks thereafter) or secukinumab (N=514; 300 mg at Week 0, 1, 2, 3, 4, and every 4 weeks thereafter). The last dose was at Week 44 for both treatment groups. Demographic and disease characteristics were similar between the two treatment groups and consistent with those of the subjects enrolled in the pivotal Phase 3 psoriasis studies for TREMFYA and secukinumab. The primary endpoint was the proportion of subjects who achieved a PASI 90 response at Week 48. Major secondary endpoints were the proportion of subjects who achieved a PASI 75 response at both Week 12 and Week 48, a PASI 90 response at Week 12, PASI 75 response at Week 12, a PASI 100 response at Week 48, an IGA score of cleared (0) at Week 48, and an IGA score of cleared (0) or minimal (1) at Week 48.
TREMFYA was superior to secukinumab as measured by the primary endpoint of PASI 90 response at Week 48 (84.5% versus 70.0%, p < 0.001). Comparative clinical response rates are presented in Table 7. (See Table 7.)
Click on icon to see table/diagram/image
TREMFYA and secukinumab PASI 90 response rates through Week 48 are presented in Figure 5. (See Figure 5.)
Click on icon to see table/diagram/image
Clinical efficacy - Psoriatic arthritis (PsA): The safety and efficacy of TREMFYA were assessed in 1120 patients in 2 randomized, double-blind, placebo-controlled studies (DISCOVER 1 and DISCOVER 2) in adult patients with active PsA (≥3 swollen joints, ≥3 tender joints, and a C-reactive protein (CRP) level of ≥0.3 mg/dL in DISCOVER 1 and ≥5 swollen joints, ≥5 tender joints, and a CRP level of ≥0.6 mg/dL in DISCOVER 2) who had inadequate response to standard therapies (e.g., conventional synthetic DMARDs [csDMARDs]), apremilast, or nonsteroidal anti-inflammatory drugs [NSAIDs]). Patients in these studies had a diagnosis of PsA for at least 6 months based on the Classification Criteria for Psoriatic Arthritis (CASPAR) and a median duration of PsA of 4 years at baseline.
In DISCOVER 1 approximately 30% of subjects had been previously treated with up to 2 anti-tumor necrosis factor alpha (anti-TNFα) agents whereas in DISCOVER 2 all subjects were biologic naïve. Approximately 58% of subjects from both studies had concomitant methotrexate (MTX) use. Patients with different subtypes of PsA were enrolled in both studies, including polyarticular arthritis with the absence of rheumatoid nodules (40%), spondylitis with peripheral arthritis (30%), asymmetric peripheral arthritis (23%), distal interphalangeal involvement (7%) and arthritis mutilans (<1%). At baseline, over 65% and 42% of the patients had enthesitis and dactylitis, respectively and over 75% had ≥3% body surface area (BSA) psoriasis skin involvement.
DISCOVER 1 evaluated 381 subjects who were treated with placebo SC, TREMFYA 100 mg SC at Weeks 0, 4 and every 8 weeks (q8w) thereafter, or TREMFYA 100 mg SC every 4 weeks (q4w). DISCOVER 2 evaluated 739 subjects who were treated with placebo SC, TREMFYA 100 mg SC at Weeks 0, 4 and q8w thereafter, or TREMFYA 100 mg SC q4w. At Week 24, placebo subjects in both studies crossed over to receive TREMFYA 100 mg SC q4w. The primary endpoint in both studies was the percentage of patients achieving an ACR20 response at Week 24. Secondary endpoints included change from baseline in Disability Index of the Health Assessment Questionnaire (HAQ-DI), IGA, ACR 50, ACR 70, SF-36 PCS, SF-36 MCS and change from baseline in total modified van der Heijde-Sharp score (DISCOVER 2), at Week 24. Additionally, resolution of enthesitis and dactylitis based on the pooled data from DISCOVER 1 and DISCOVER 2 was assessed as a secondary endpoint in DISCOVER 2.
Signs and symptoms: In both studies, patients treated with TREMFYA 100 mg q8w or 100 mg q4wW demonstrated a greater clinical response including ACR20, ACR50, and ACR70 compared to placebo at Week 24 (Table 8). These responses were maintained from Week 24 to Week 52. Responses were seen regardless of prior anti-TNFα exposure (DISCOVER 1) and concomitant csDMARD use (DISCOVER 1 and DISCOVER 2). Additionally, in both studies, examination of age, gender, race, body weight, and previous treatment with csDMARDs did not identify differences in response to TREMFYA among these subgroups. (See Table 8.)
Click on icon to see table/diagram/image
In DISCOVER 1 and 2, patients treated with TREMFYA who had spondylitis with peripheral arthritis as their primary presentation, demonstrated greater improvement from baseline in Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) compared to placebo at Week 24. In both studies, the improvement in BASDAI was maintained from Week 24 to Week 52.
In DISCOVER 2, a greater ACR 20 response was observed in both TREMFYA dose groups compared with the placebo group as early as Week 4 and the treatment difference continued to increase over time through Week 24 (Figure 6). (See Figure 6.)
Click on icon to see table/diagram/image
ACR 20 response was maintained from Week 24 to Week 52.
In DISCOVER 1 and DISCOVER 2, improvements as assessed by mean change from baseline were observed in all components of the ACR response criteria (see Table 9).
Click on icon to see table/diagram/image
Psoriasis Skin Response: In DISCOVER 1 and DISCOVER 2, among subjects with mild to severe psoriasis (BSA ≥3% and IGA ≥2) at baseline, a greater proportion of subjects in both TREMFYA dose groups achieved a psoriasis response, defined as an IGA of 0 (cleared) or 1 (minimal) and a ≥2-grade reduction from baseline, compared with the placebo group at Week 24. Results from psoriasis skin response endpoints in DISCOVER 1 and DISCOVER 2 are presented in Table 10. Responses for both IGA and PASI endpoints were maintained from Week 24 to Week 52. (See Table 10.)
Click on icon to see table/diagram/image
Enthesitis and Dactylitis: Enthesitis and dactylitis were assessed based on pooled data from DISCOVER 1 and DISCOVER 2. Among subjects with dactylitis at baseline, a greater proportion of subjects in both the TREMFYA 100 mg q8w and the TREMFYA 100 mg q4w groups achieved dactylitis resolution at Week 24 compared with the placebo group (Table 11). Among subjects with enthesitis at baseline, a greater proportion of subjects in both the TREMFYA 100 mg q8w group and q4w group achieved enthesitis resolution at Week 24 compared with the placebo group (Table 11). Based on the combined data from DISCOVER 1 and DISCOVER 2, resolution of dactylitis and enthesitis were maintained from Week 24 to Week 52. (See Table 11.)
Click on icon to see table/diagram/image
Radiographic response: In DISCOVER 2, inhibition of structural damage progression was measured radiographically and expressed as the mean change from baseline in the total modified van der Heijde-Sharp (vdH-S) score.
At Week 24, the guselkumab q4w group demonstrated statistically significantly less radiographic progression and the guselkumab q8w group showed numerically less progression than placebo (Table 12). The observed benefit with the guselkumab q4w dosing regimen on inhibition of radiographic progression (ie, smaller mean change from baseline in total modified vdH-S score in the q4w group versus placebo) was most pronounced in subjects with both a high C-reactive protein value and high number of joints with erosions at baseline. (See Table 12.)
Click on icon to see table/diagram/image
At Week 52, the mean change from baseline in total modified vdH-S was similar in the guselkumab q8w and q4w groups (Table 13). (See Table 13.)
Click on icon to see table/diagram/image
Physical function and health-related quality of life: TREMFYA-treated patients in both the 100 mg q8w and q4w dose groups in both DISCOVER 1 and DISCOVER 2 showed greater mean improvement from baseline in physical function compared to patients treated with placebo as assessed by HAQ-DI at Weeks 16 and 24. Improvements in HAQ-DI were maintained from Week 24 to Week 52. In both studies, the proportion of HAQ-DI responders (≥ 0.35 improvement in HAQ-DI score) was greater in both TREMFYA dose groups compared to placebo at weeks 16 and 24. The proportion of HAQ-DI responders was maintained from Week 24 to Week 52. (SeeTable 14.)
Click on icon to see table/diagram/image
At Week 24, subjects in both the TREMFYA 100 mg q8w and q4w dose groups in both DISCOVER 1 and DISCOVER 2 showed greater improvement from baseline in the SF-36 PCS with no worsening in the SF-36 MCS compared with placebo. At Week 24 there was consistent evidence of effect in the physical functioning, role-physical, bodily-pain, general health, social-functioning and vitality domains but not in the role-emotional and mental health domains. Subjects in both the TREMFYA 100 mg q8w and q4w dose groups in both DISCOVER 1 and DISCOVER 2 showed greater improvement compared with placebo in fatigue measured with FACIT-fatigue at Week 24. In DISCOVER 2, greater improvements in health-related quality of life as measured by the Dermatology Life Quality Index (DLQI) were observed in guselkumab-treated patients compared to placebo at Week 24. In DISCOVER 2, greater improvements were also observed in overall work impairment and activity impairment as assessed by the Work Productivity and Activity Impairment (WPAI)-PsA questionnaire compared to placebo at Week 24. Improvements in SF-36 PCS, SF-36 MCS, FACIT-F, DLQI and WPAI-PsA scores were maintained from Week 24 to 52. (See Table 15.)
Click on icon to see table/diagram/image
Clinical efficacy-palmoplantar pustulosis (PPP): Study CNTO1959PPP3001 was a double-blind, placebo-controlled study which evaluated the efficacy and safety of TREMFYA in 159 Japanese subjects with PPP. The study enrolled subjects who had been diagnosed with PPP at least 24 weeks prior to screening; had an inadequate response to topical steroids, topical vitamin D3 derivative preparations, systemic etretinate, or phototherapy; and had a palmoplantar pustulosis area and severity index (PPPASI) total score of at least 12 and a PPPASI severity score of at least 2 for pustules and vesicles on the palms or soles. Subjects were randomized to receive TREMFYA 100 mg (recommended dose), TREMFYA 200 mg or placebo at Weeks 0 and 4, and every 8 weeks thereafter. Subjects randomized to placebo were re-randomized to receive TREMFYA 100 mg or 200 mg at Weeks 16, 20 and every 8 weeks thereafter.
The primary endpoint was the change in total PPPASI score from baseline at Week 16. Other endpoints included the proportion of subjects who achieved PPPASI-50 response at Week 16, and the change from baseline in Palmoplantar Pustulosis Severity Index (PPSI) at Week 16.
The change from baseline in PPPASI score at Week 16 was significantly higher in the TREMFYA 100 mg group compared to the placebo group. PPPASI-50 response in the TREMFYA 100 mg group was 57.4% (31/54 patients) at Week 16 and 83.3% (45/54 patients) at Week 52, respectively. No incremental benefit was observed with the TREMFYA 200 mg dose. (See Table 16.)
Click on icon to see table/diagram/image
Pharmacokinetics: Absorption: Following a single 100 mg subcutaneous injection in healthy subjects, guselkumab reached a mean (± SD) maximum serum concentration (C
max) of 8.09 ± 3.68 mcg/mL by approximately 5.5 days post dose.
Steady-state serum guselkumab concentrations were achieved by Week 20 following subcutaneous administrations of 100 mg guselkumab at Weeks 0 and 4, and every 8 weeks thereafter. The mean (± SD) steady-state trough serum guselkumab concentrations in two Phase 3 studies were 1.15 ± 0.73 mcg/mL and 1.23 ± 0.84 mcg/mL. Serum guselkumab concentrations did not appear to accumulate over time when given subcutaneously every 8 weeks.
The pharmacokinetics of guselkumab in subjects with psoriatic arthritis was similar to that in subjects with plaque psoriasis. Following subcutaneous administration of 100 mg of guselkumab at Weeks 0, 4, and every 8 weeks thereafter, mean steady-state trough serum guselkumab concentration was approximately 1.2 mcg/mL. Following subcutaneous administration of 100 mg of guselkumab every 4 weeks, mean steady-state trough serum guselkumab concentration was approximately 3.8 mcg/mL.
The absolute bioavailability of guselkumab following a single 100 mg subcutaneous injection was estimated to be approximately 49% in healthy subjects.
Distribution: Mean volume of distribution during the terminal phase (Vz) following a single intravenous administration to healthy subjects ranged from approximately 7 to 10 L (98 to 123 mL/kg) across studies.
Metabolism: The exact pathway through which guselkumab is metabolized has not been characterized. As a human IgG monoclonal antibody, guselkumab is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG.
Elimination: Mean systemic clearance (CL) following a single intravenous administration to healthy subjects ranged from 0.288 to 0.479 L/day (3.6 to 6.0 mL/day/kg) across studies.
Mean half-life (T
½) of guselkumab was approximately 17 days in healthy subjects and approximately 15 to 18 days in subjects with plaque psoriasis across studies.
Dose linearity: The systemic exposure of guselkumab (C
max and AUC) increased in an approximately dose-proportional manner following a single subcutaneous injection at doses ranging from 10 mg to 300 mg in healthy subjects or subjects with plaque psoriasis.
Population pharmacokinetic analysis: In a population pharmacokinetic analysis, the apparent clearance (CL/F) and apparent volume of distribution (V/F) were 0.516 L/d and 13.5 L, respectively, and the T
½ was approximately 18 days in subjects with psoriasis.
In the population pharmacokinetic analysis, the effects of baseline demographics (weight, age, sex, and race), immunogenicity, baseline disease characteristics, comorbidities (past and current history of diabetes, hypertension, and hyperlipidemia), past use of therapeutic biologics, past use of methotrexate or cyclosporine, concomitant medications (NSAIDs, corticosteroids and conventional synthetic DMARDS such as methotrexate), use of alcohol, or current smoking status, on pharmacokinetics of guselkumab was evaluated. Only the effects of body weight on CL/F and V/F were found to be significant, with a trend towards higher CL/F in heavier subjects. However, subsequent exposure-response modeling analysis suggested that no dose adjustment would be warranted for body weight.
Cytochrome P450 Substrates: An
in vitro study using human hepatocytes showed that IL-23 did not alter the expression or activity of multiple CYP450 enzymes (CYP1A2, 2B6, 2C9, 2C19, 2D6, or 3A4).
The effects of guselkumab on the pharmacokinetics of representative probe substrates of CYP isozymes (midazolam [CYP3A4], warfarin [CYP2C9], omeprazole [CYP2C19], dextromethorphan [CYP2D6], and caffeine [CYP1A2]) were evaluated in subjects with moderate to severe plaque psoriasis. Results from this study indicate that changes in C
max and AUC
inf of midazolam, S-warfarin, omeprazole, dextromethorphan, and caffeine after a single dose of guselkumab were not clinically relevant (see Interactions).
There is no need for dose adjustment when co-administering guselkumab and CYP450 substrates.
Special populations: Pediatrics (17 years of age and younger): The safety and efficacy of guselkumab have not been established in pediatric patients.
Elderly (65 years of age and older): Of the 1384 plaque psoriasis subjects exposed to TREMFYA in Phase 3 clinical studies and included in the population pharmacokinetic (pop PK) analysis, 70 subjects were 65 years of age or older, including 4 subjects who were 75 years of age or older. Population pharmacokinetic analyses indicated there were no apparent changes in CL/F estimate in subjects ≥ 65 years of age compared to subjects < 65 years of age, suggesting no dose adjustment is needed for elderly patients. Of the 746 psoriatic arthritis patients exposed to TREMFYA in Phase 3 clinical studies and included in the pop PK analysis, a total of 38 patients were 65 years of age or older, and no patients were 75 years of age or older.
Renal impairment: No specific study has been conducted to determine the effect of renal impairment on the pharmacokinetics of guselkumab.
Hepatic impairment: No specific study has been conducted to determine the effect of hepatic impairment on the pharmacokinetics of guselkumab.
Toxicology: Non-Clinical Information: In repeat-dose toxicity studies in cynomolgus monkeys, guselkumab was well-tolerated at weekly doses up to 50 mg/kg intravenously for 5 weeks or 50 mg/kg subcutaneously for up to 24 weeks. There were no effects on cardiovascular, respiratory and nervous system function, and clinical pathology or anatomical pathology parameters. Safety margins at the NOAEL dose (50 mg/kg once weekly) were approximately 206-fold and 50-fold higher for AUC
last and C
max, respectively, than those following a single administration of a 100 mg SC dose to psoriasis subjects.
Carcinogenicity and Mutagenicity: Routine genotoxicity and carcinogenicity studies were not performed as large proteins cannot diffuse into cells and cannot interact with DNA or chromosomal material.
Reproductive Toxicology: There were no effects on reproduction or development in a prenatal and postnatal developmental toxicity (ePPND) study in which pregnant cynomolgus monkeys were administered guselkumab SC at doses up to 50 mg/kg/week from gestation day 20 through natural delivery. Peak serum concentrations in pregnant monkeys were 152-fold and 36-fold higher for C
max and AUC, respectively than those observed in psoriasis subjects following a single administration of a 100 mg SC dose. Guselkumab was detectable in newborn cynomolgus monkey serum samples indicating transplacental transfer of drug. Guselkumab was undetectable in breast milk at 4 weeks postpartum. There was a slightly higher incidence of pregnancy losses in the guselkumab treatment groups (10 or 50 mg/kg/week SC) relative to controls but without clear dose-response relationship. The clinical significance of these findings is unknown.
Immunization of infant monkeys with KLH at 4 to 6 months of age showed no impairment in the ability of the infants to mount a T-cell dependent anti-KLH antibody response to KLH immunization.
Fertility: No effects on fertility parameters were identified in female and male fertility studies conducted in guinea pigs. Results from the studies indicated no effects on male or female reproductive parameters, including no localization of guselkumab by immunohistochemistry (IHC) in any female reproductive tissues at 3 time points following mating in one mechanistic study. Safety margins for C
max and AUC
last at the 100 mg/kg twice-weekly NOAEL dose were at least 106-fold and 12-fold higher, respectively than those following a single administration of a 100 mg SC dose to psoriasis subjects.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image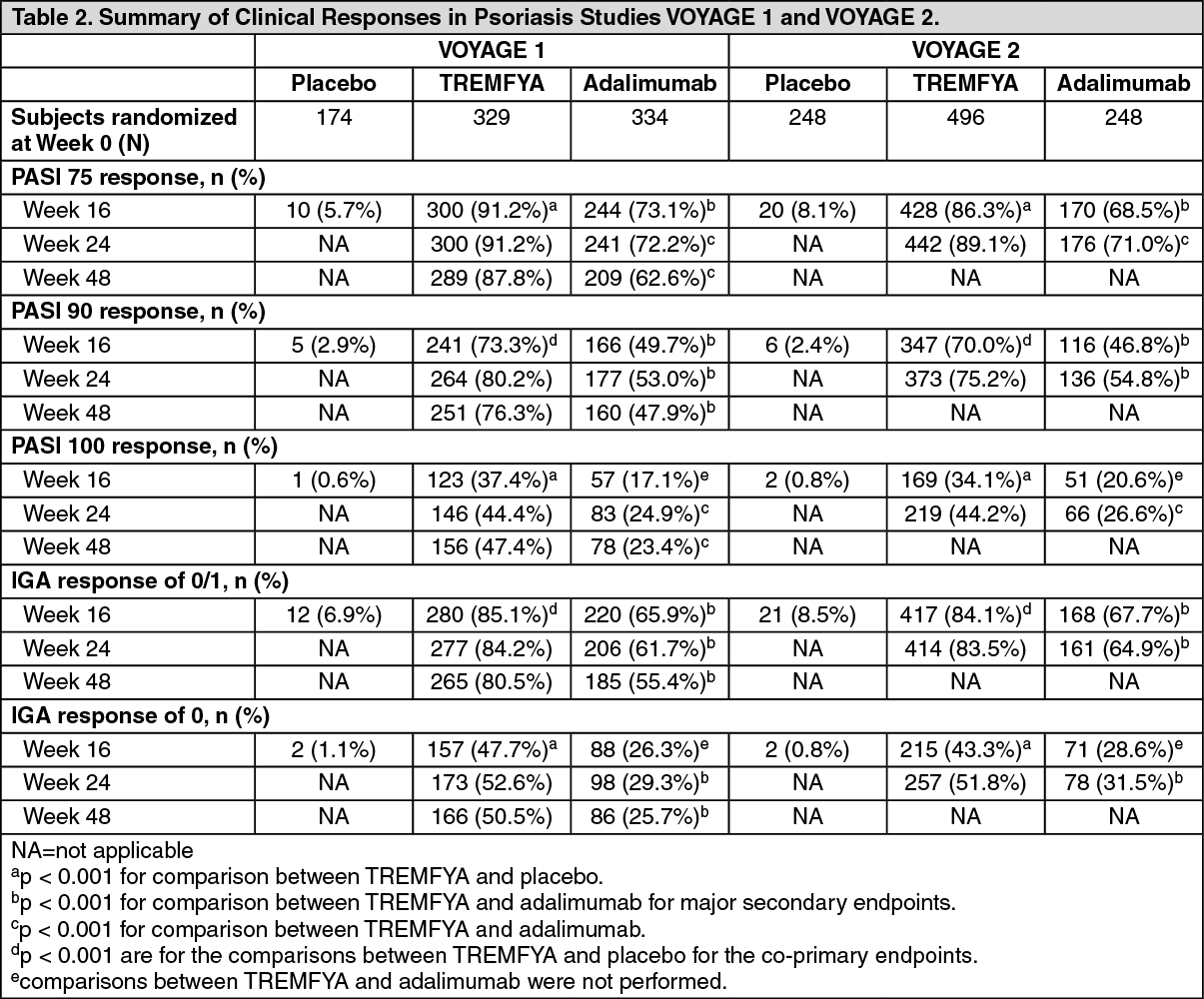 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image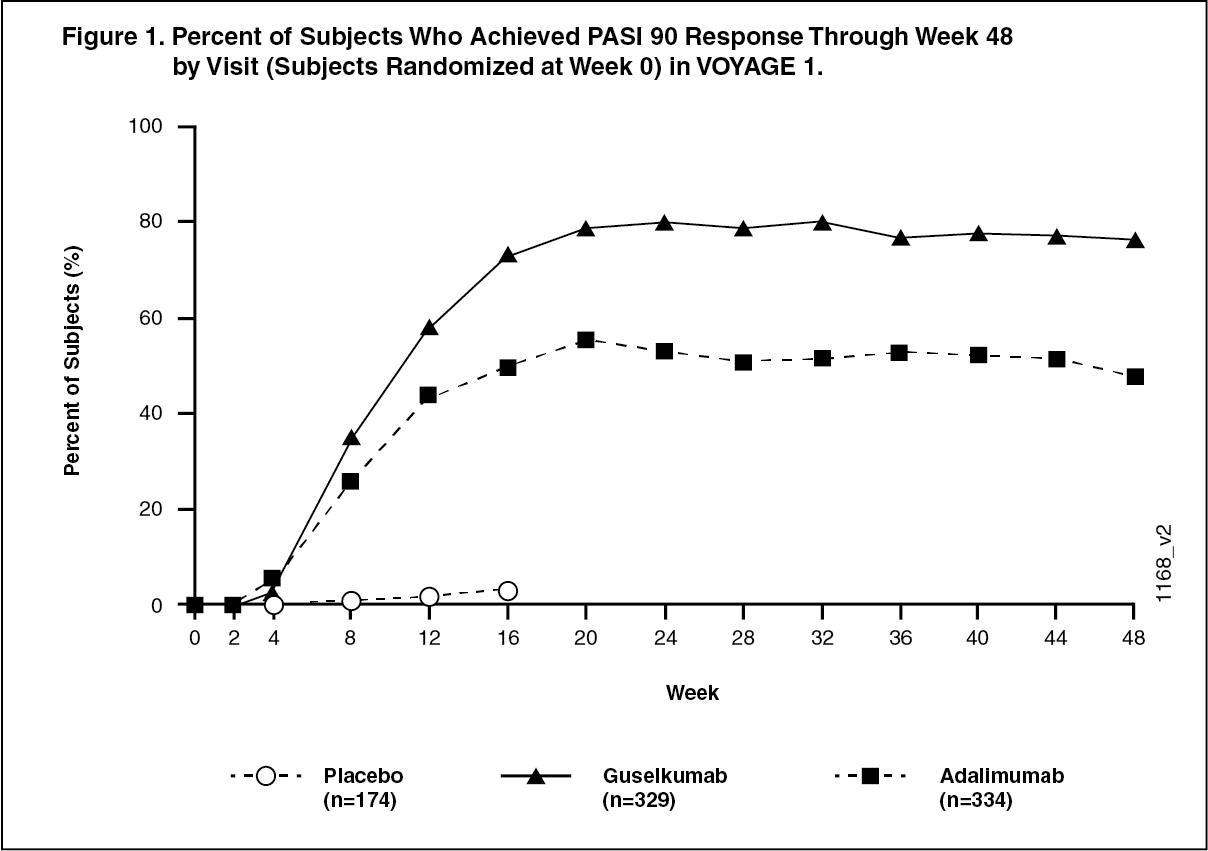 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image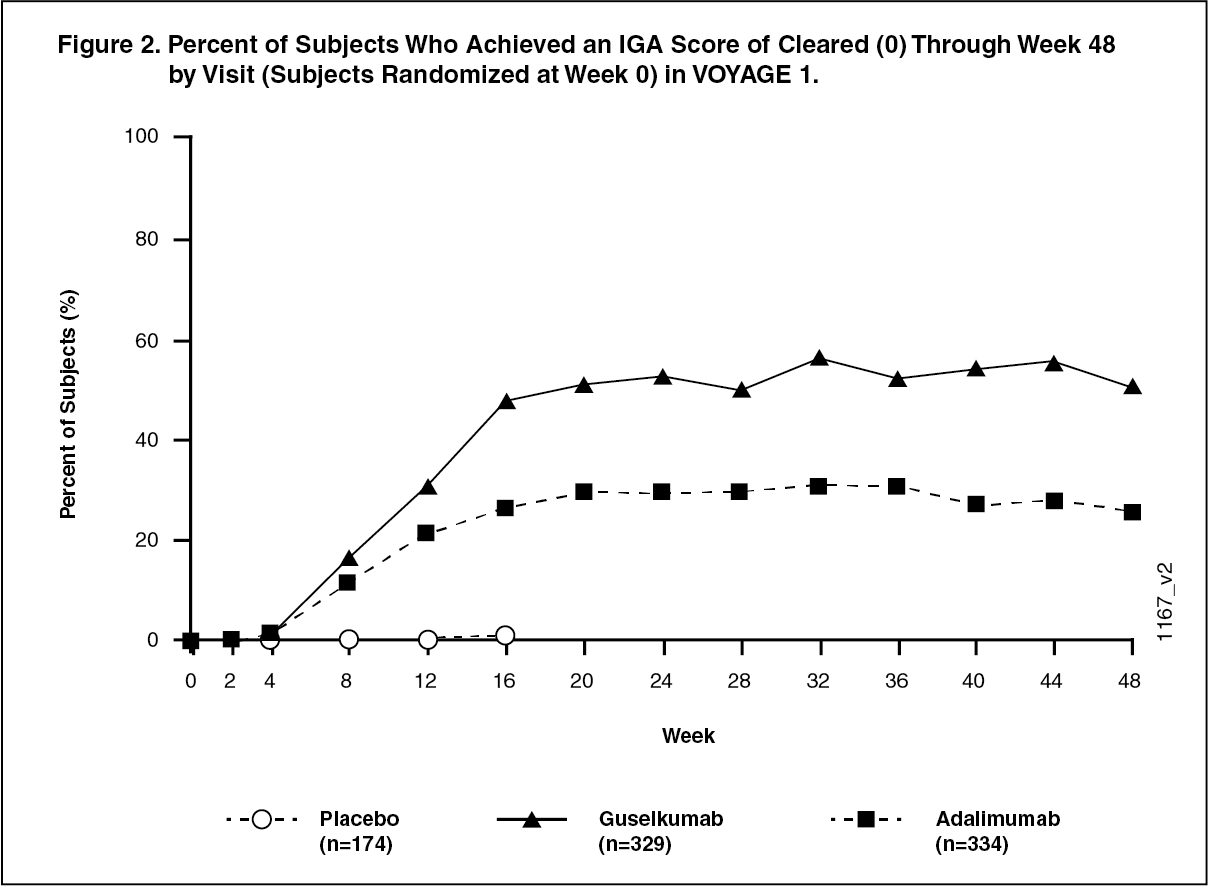 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image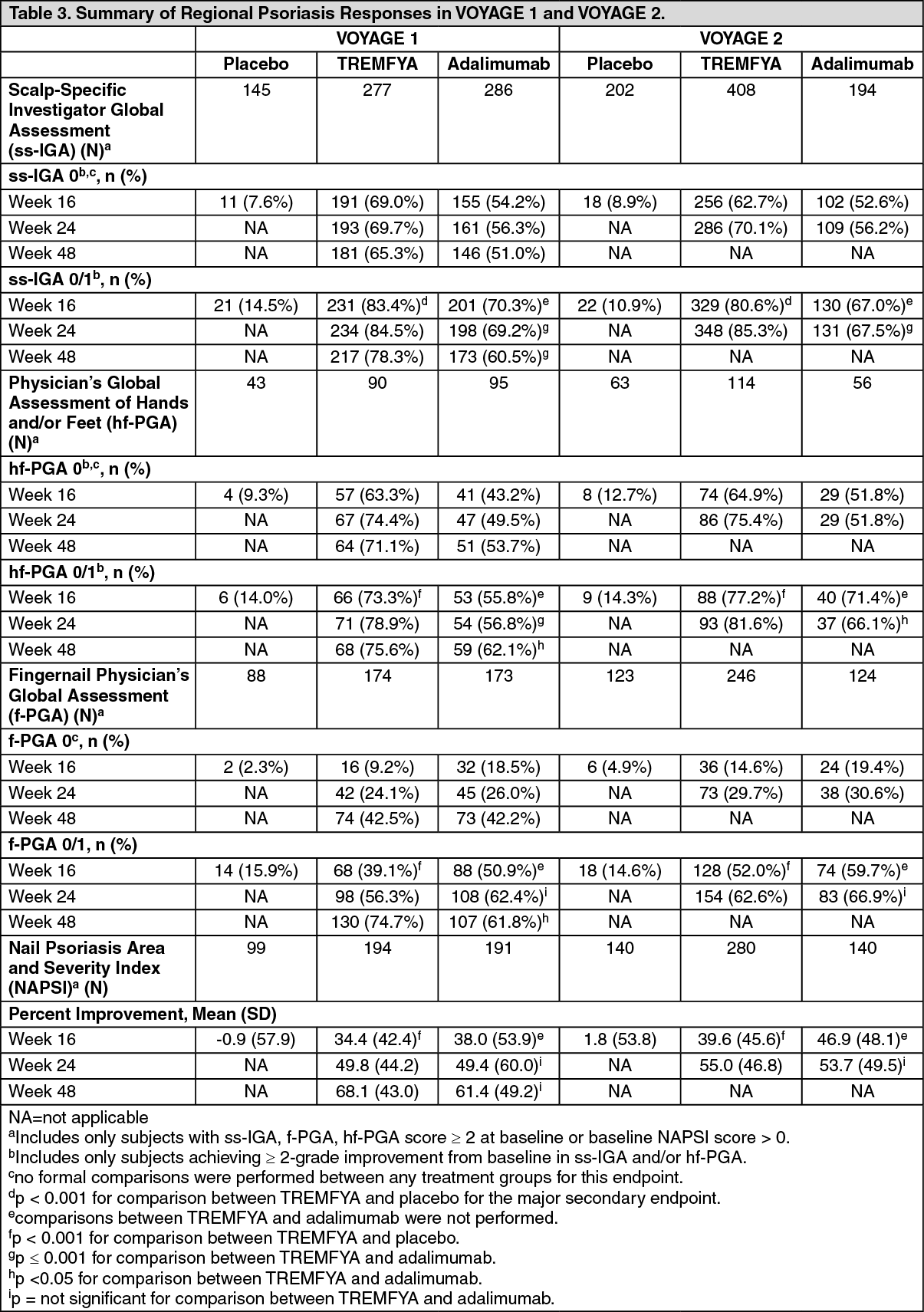 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image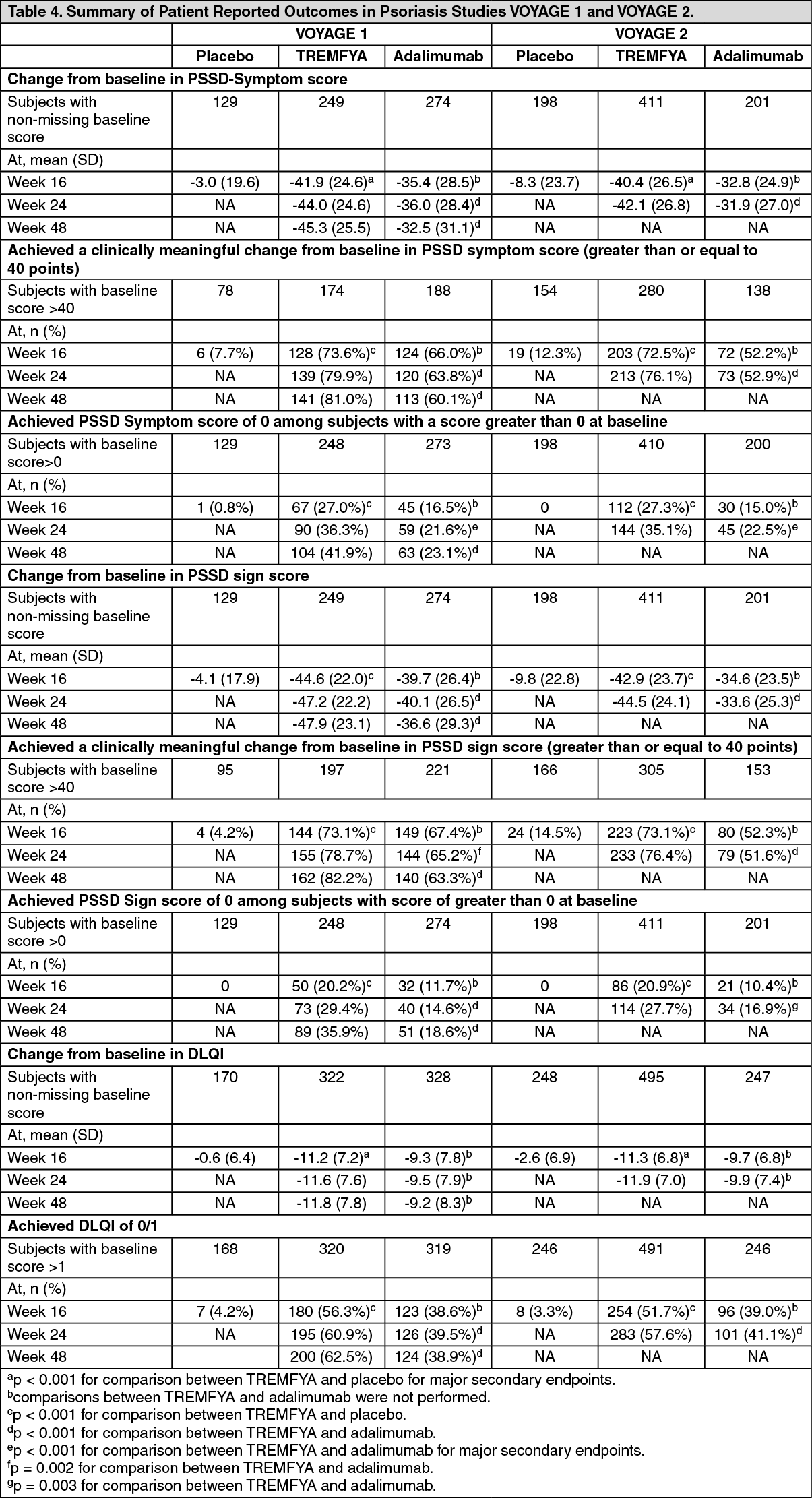 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image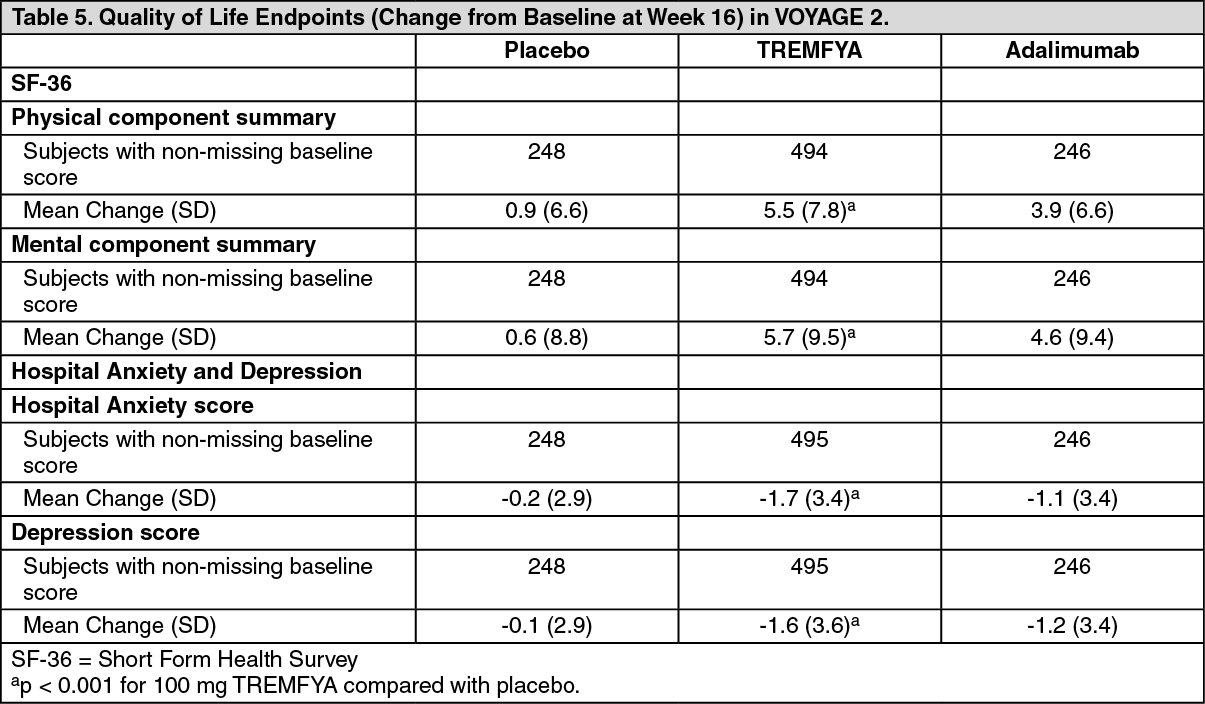 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image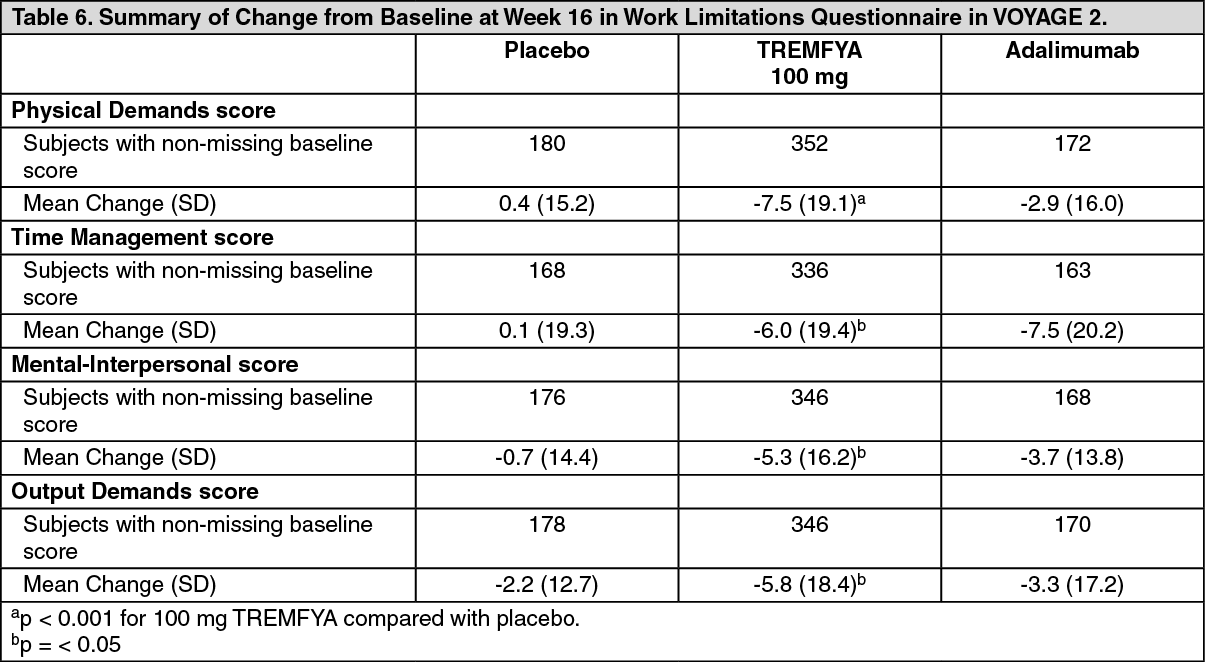 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image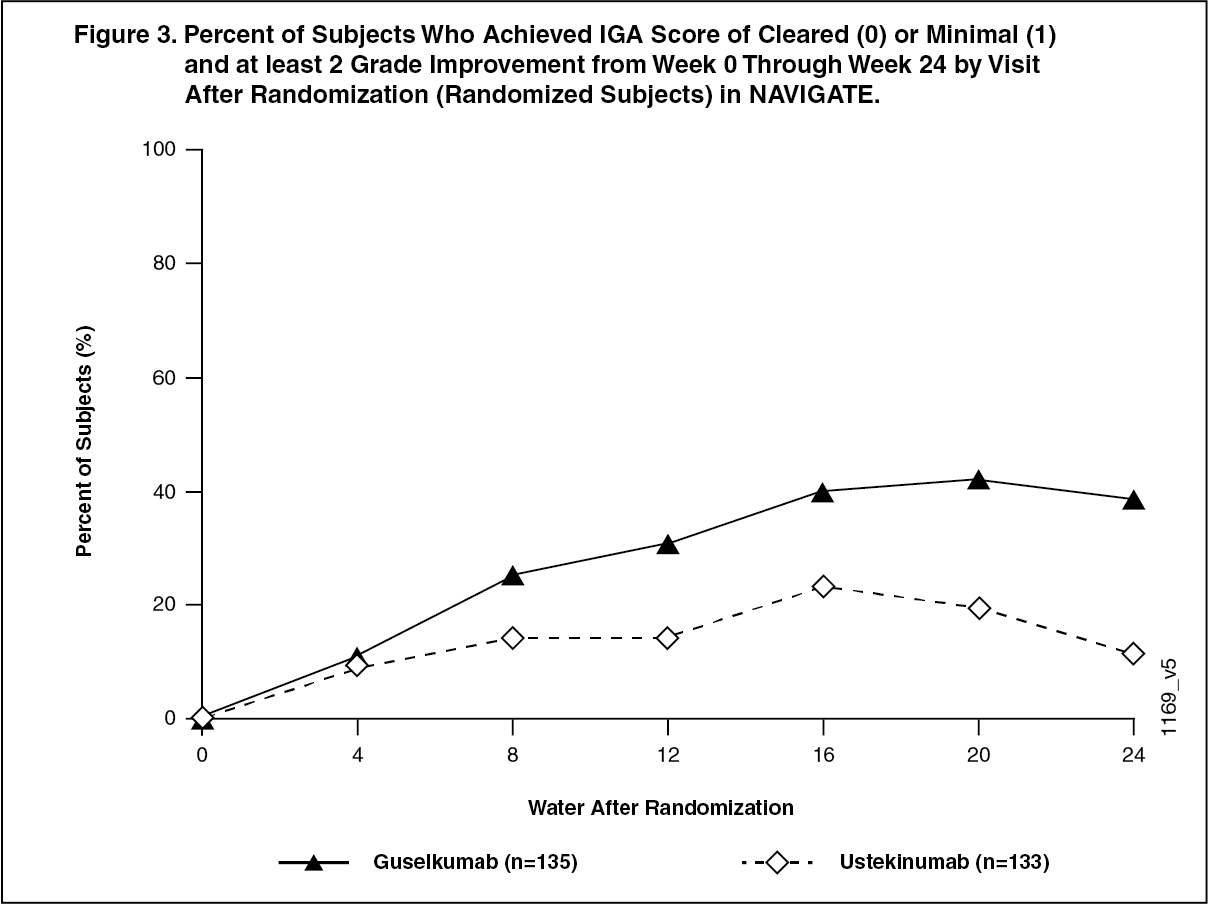 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image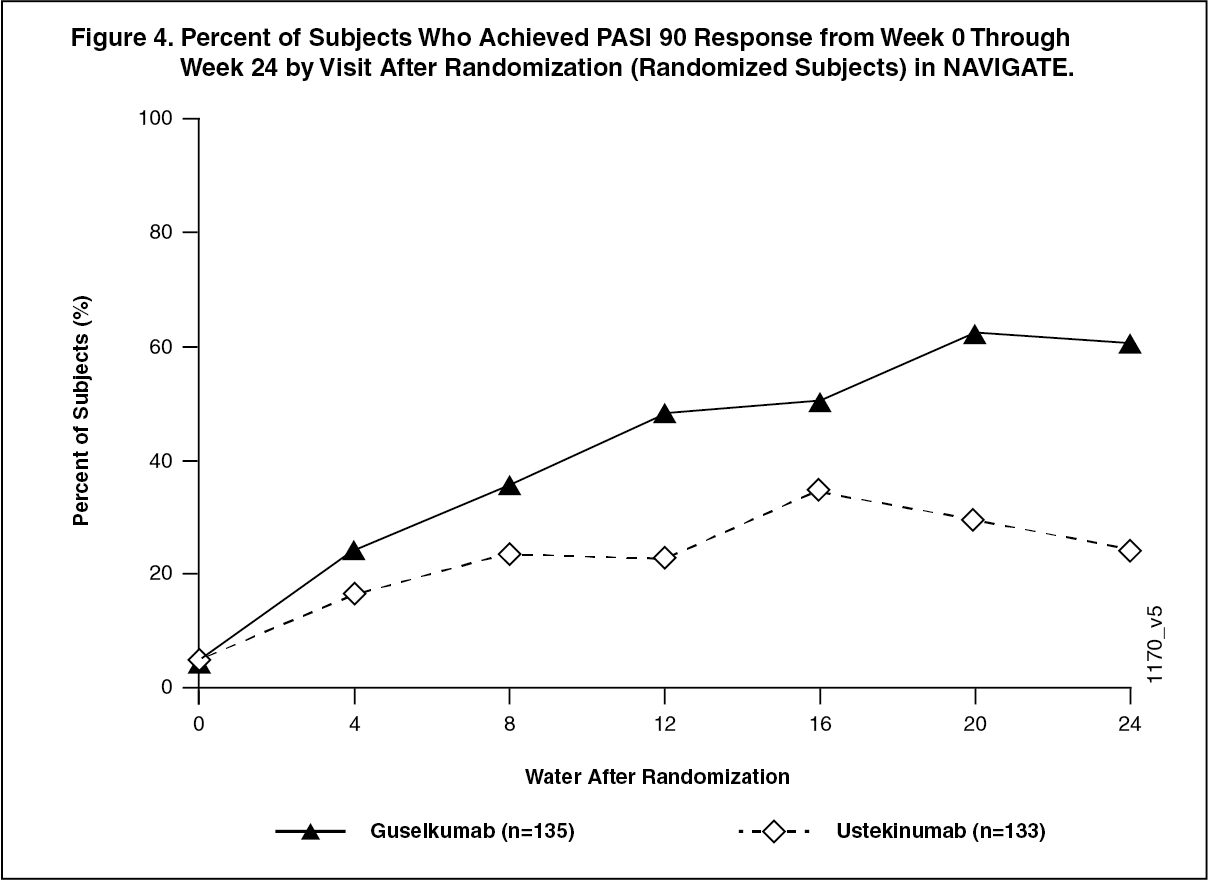 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image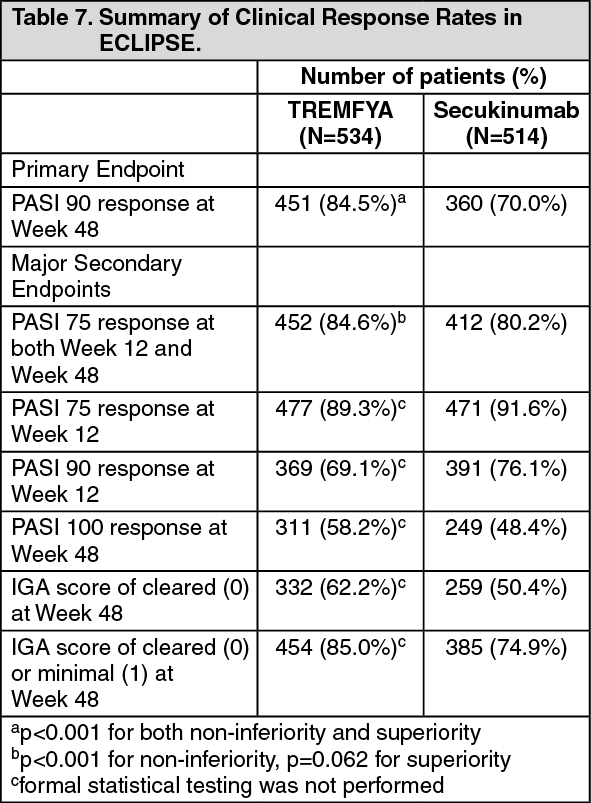 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image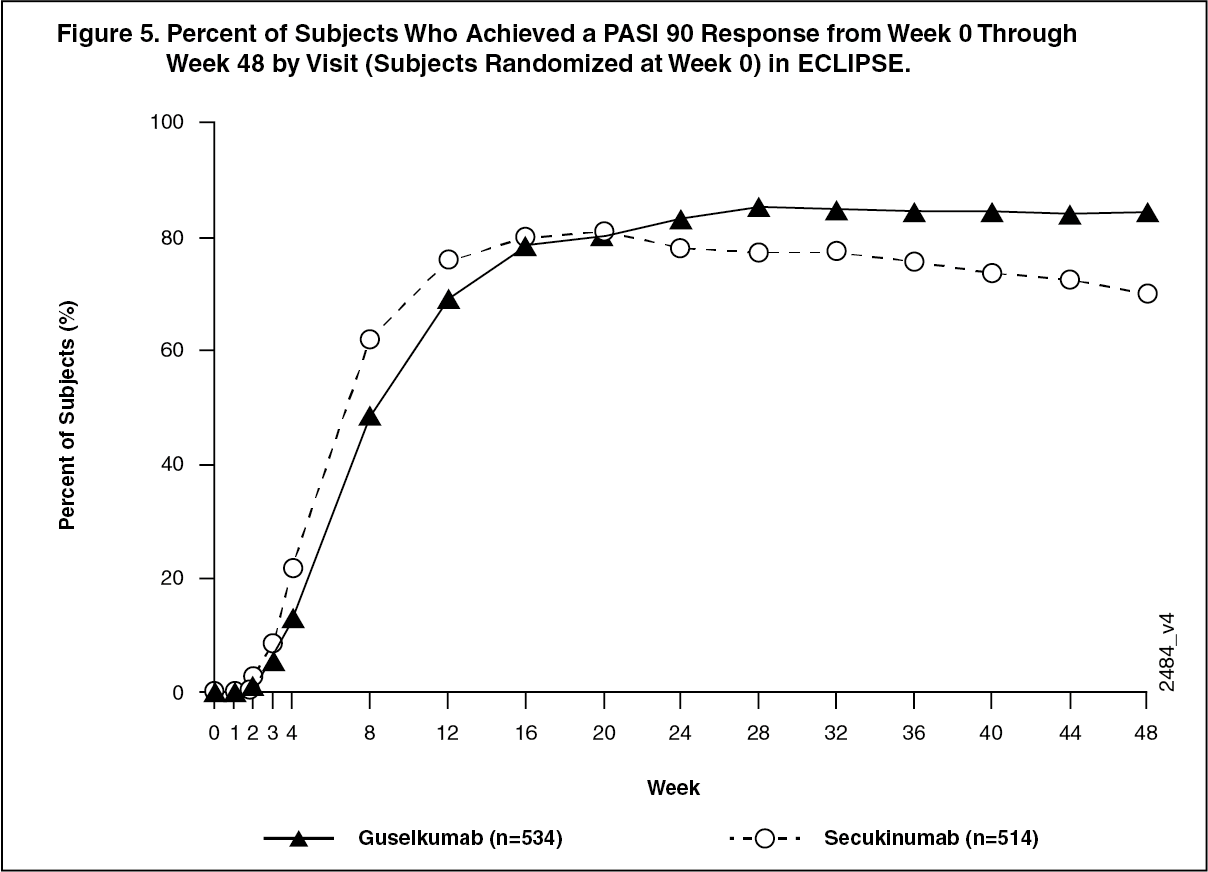 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image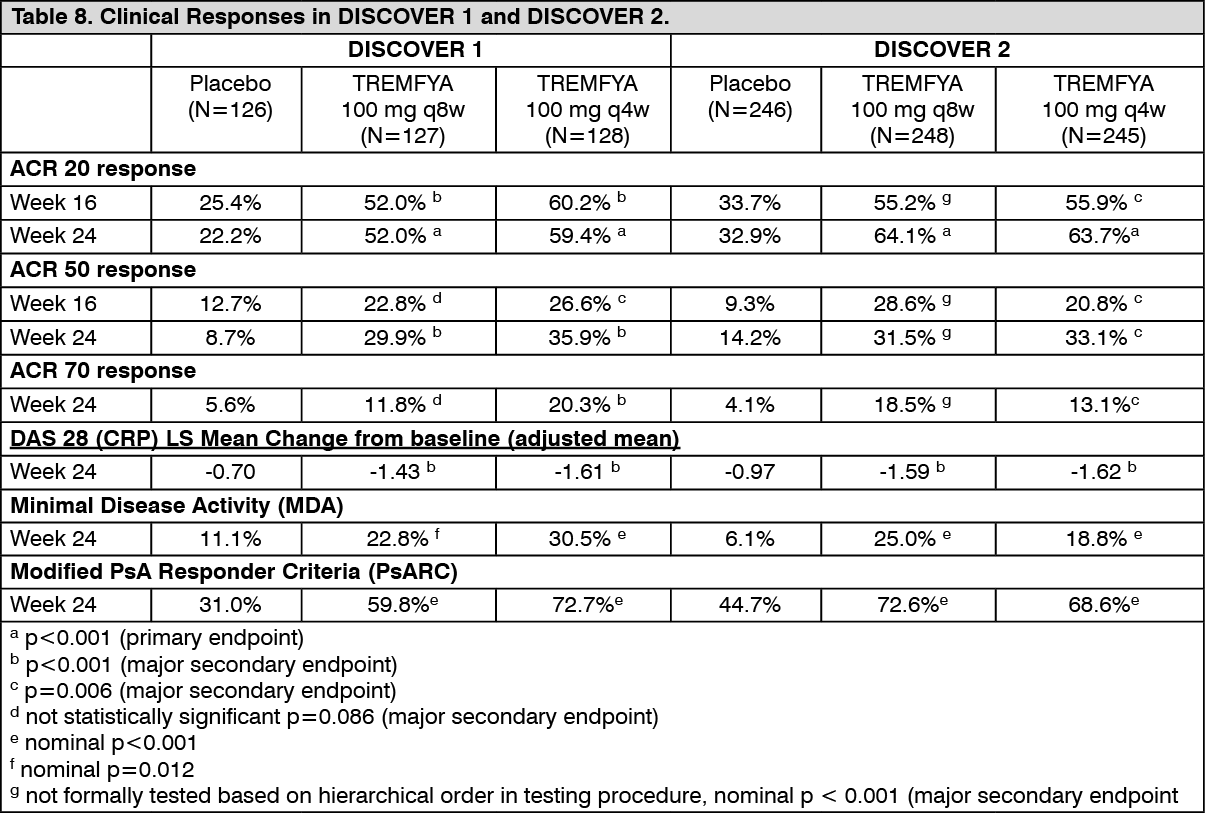 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image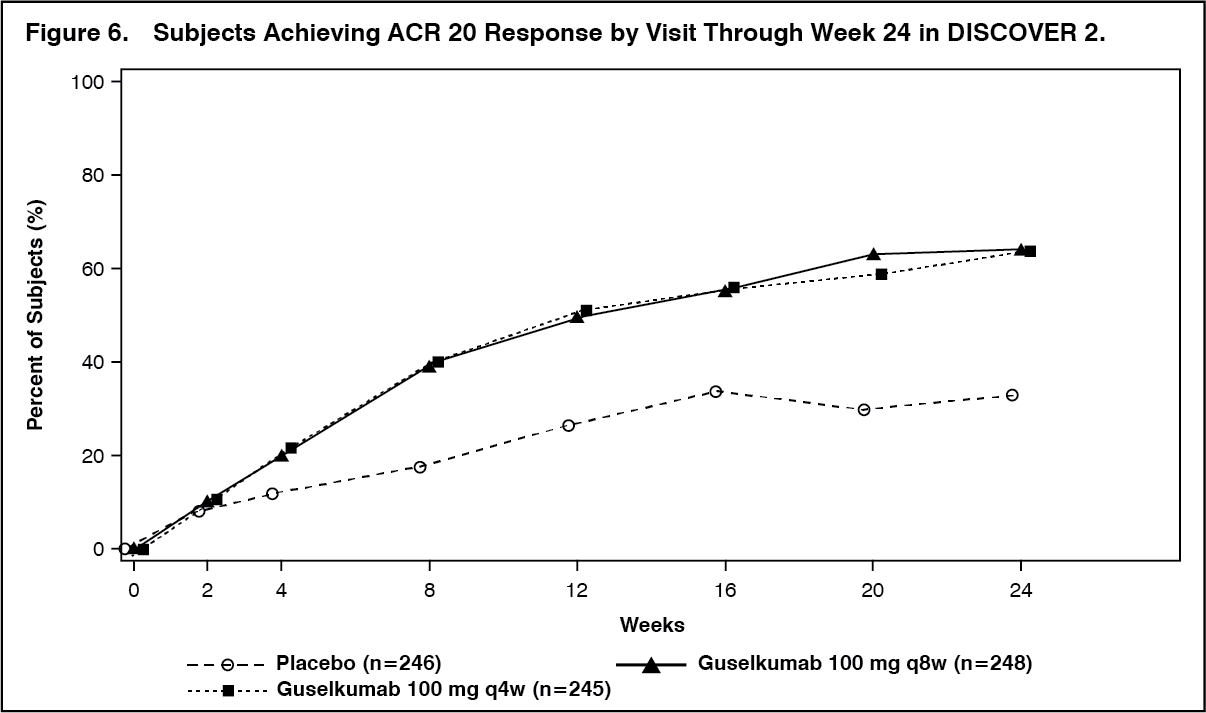 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image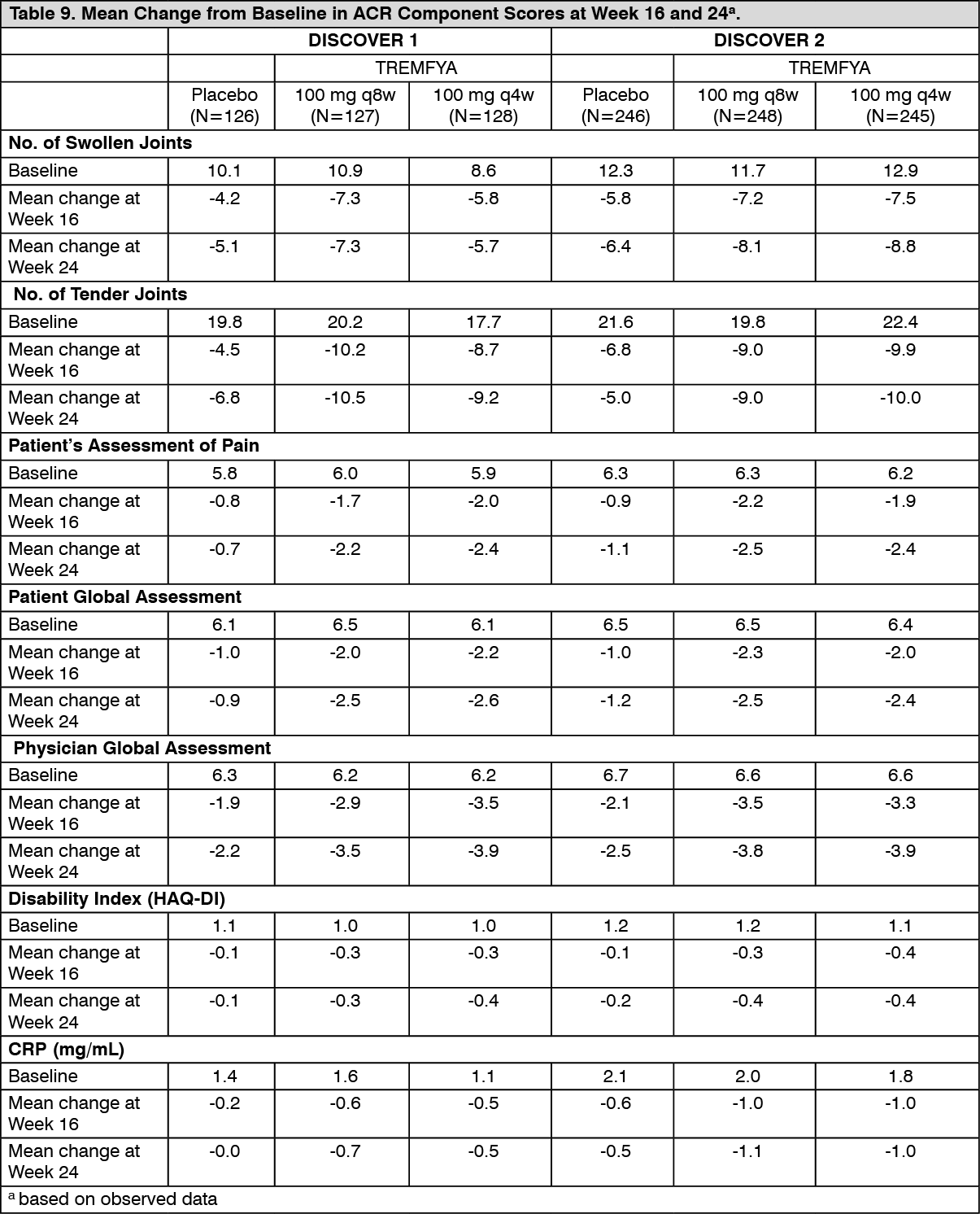 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image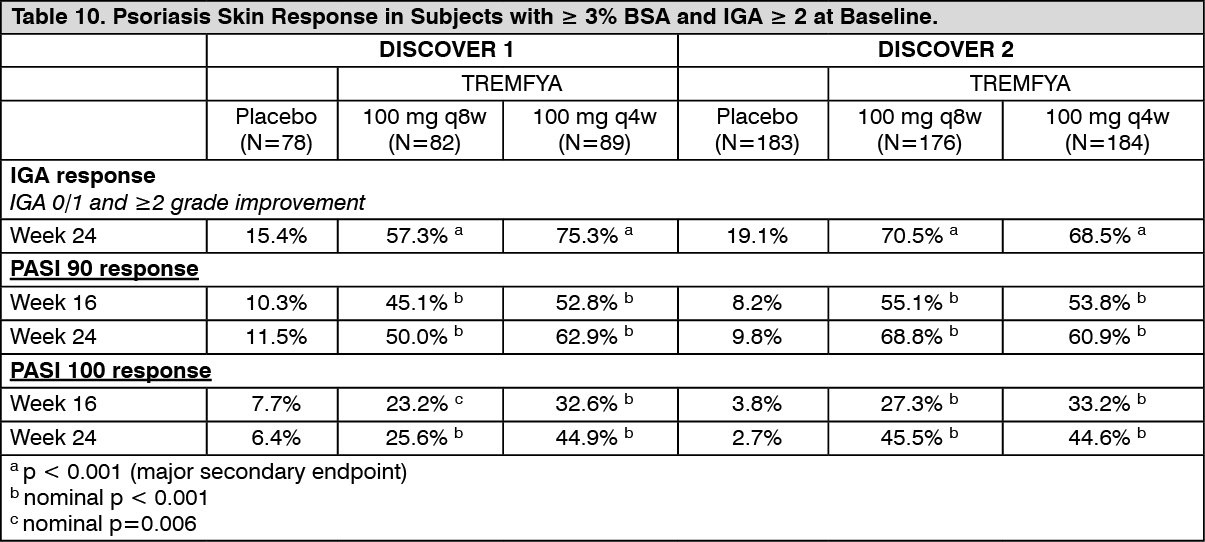 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image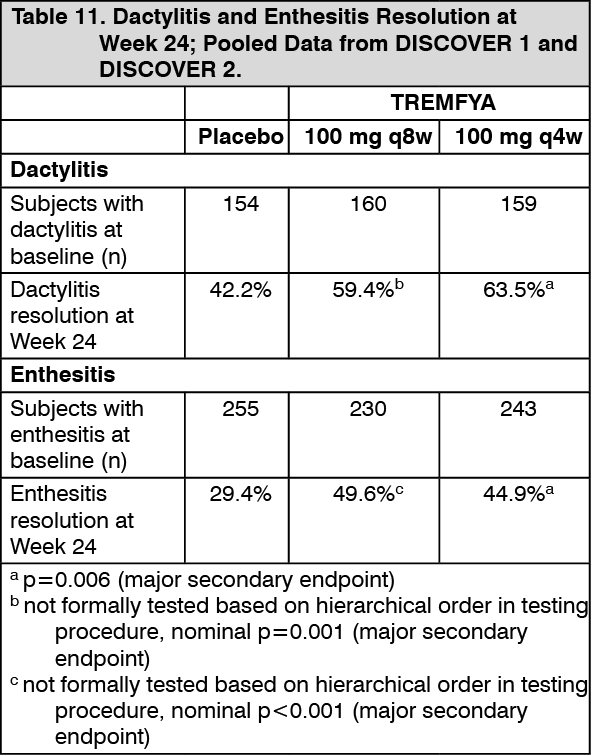 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image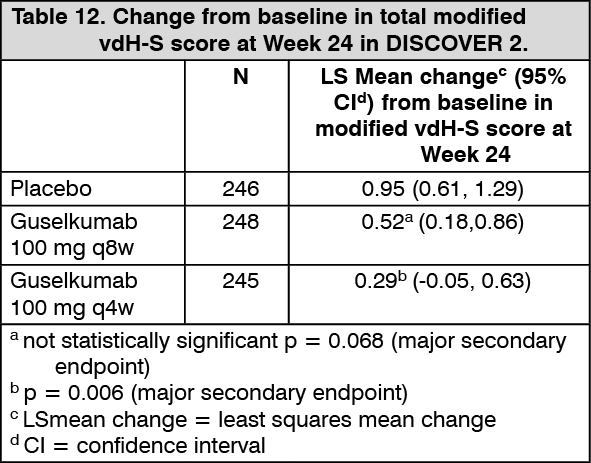 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image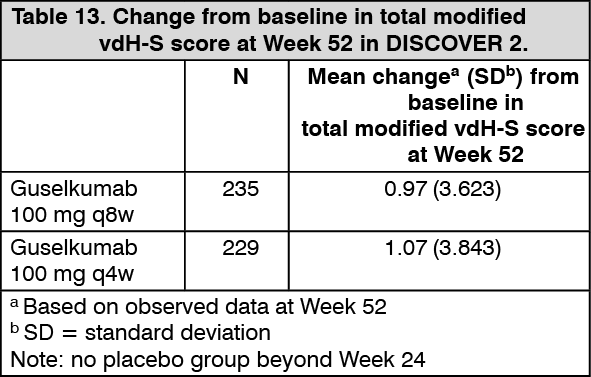 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image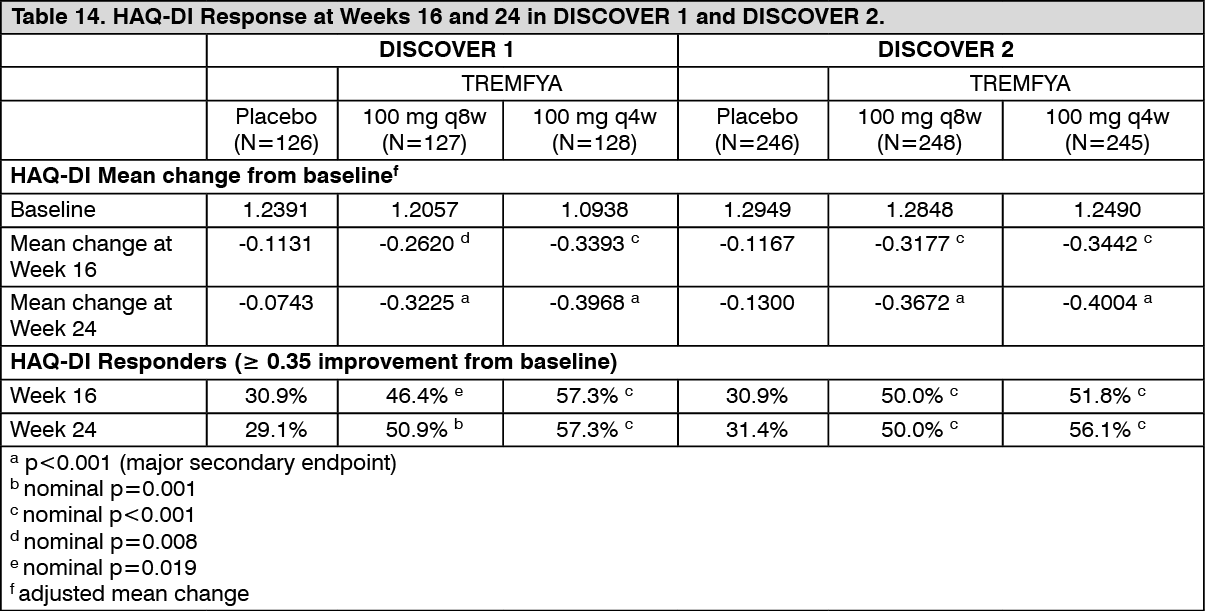 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image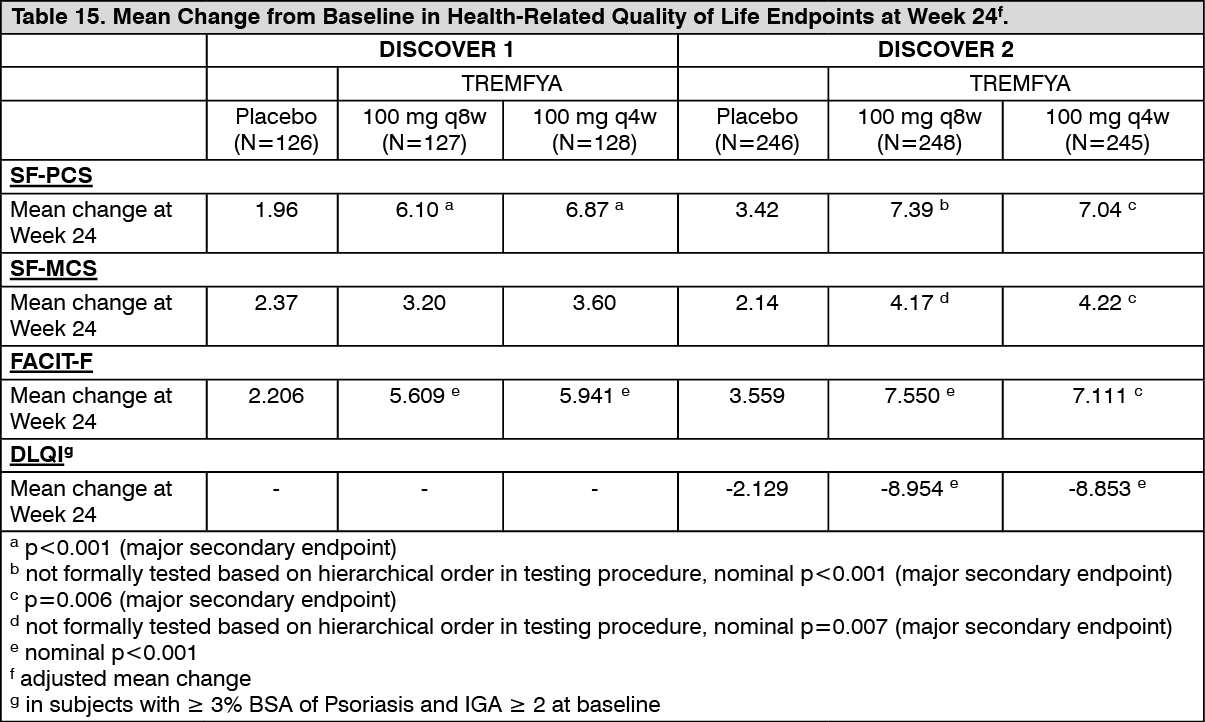 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image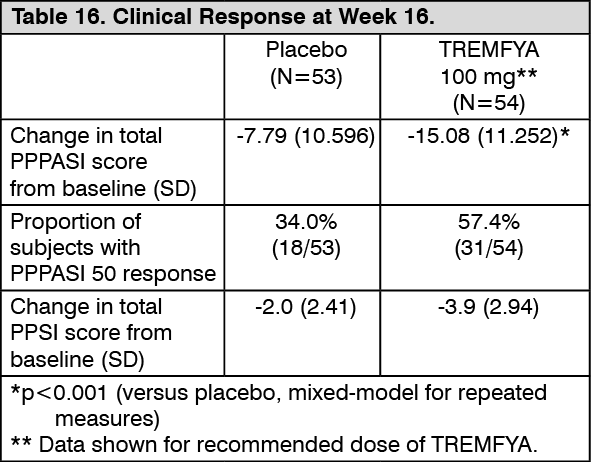 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
