


 Sign Out
Sign Out



Pharmacology: Pharmacodynamics: Mechanism of action: Bosentan is a dual endothelin receptor antagonist (ERA) with affinity for both endothelin A and B (ETA and ETB) receptors. Bosentan decreases both pulmonary and systemic vascular resistance resulting in increased cardiac output without increasing heart rate.
The neurohormone endothelin-1 (ET-1) is one of the most potent vasoconstrictors known and can also promote fibrosis, cell proliferation, cardiac hypertrophy and remodelling, and is pro-inflammatory. These effects are mediated by endothelin binding to ETA and ETB receptors located in the endothelium and vascular smooth muscle cells. ET-1 concentrations in tissues and plasma are increased in several cardiovascular disorders and connective tissue diseases, including PAH, scleroderma, acute and chronic heart failure, myocardial ischaemia, systemic hypertension and atherosclerosis, suggesting a pathogenic role of ET-1 in these diseases. In PAH and heart failure, in the absence of endothelin receptor antagonism, elevated ET-1 concentrations are strongly correlated with the severity and prognosis of these diseases.
Bosentan competes with the binding of ET-1 and other ET peptides to both ETA and ETB receptors, with a slightly higher affinity for ETA receptors (Ki = 4.1-43 nanomolar) than for ETB receptors (Ki = 38-730 nanomolar). Bosentan specifically antagonises ET receptors and does not bind to other receptors.
Efficacy: Animal models: In animal models of pulmonary hypertension, chronic oral administration of bosentan reduced pulmonary vascular resistance and reversed pulmonary vascular and right ventricular hypertrophy. In an animal model of pulmonary fibrosis, bosentan reduced collagen deposition in the lungs.
Efficacy in adult patients with pulmonary arterial hypertension: Two randomised, double-blind, multi-centre, placebo-controlled studies have been conducted in 32 (study AC-052-351) and 213 (study AC-052-352 [BREATHE-1]) adult patients with WHO functional class III-IV PAH (primary pulmonary hypertension or pulmonary hypertension secondary mainly to scleroderma). After 4 weeks of bosentan 62.5 mg twice daily, the maintenance doses studied in these studies were 125 mg twice daily in AC-052-351, and 125 mg twice daily and 250 mg twice daily in AC-052-352.
Bosentan was added to patients' current therapy, which could include a combination of anticoagulants, vasodilators (e.g., calcium channel blockers), diuretics, oxygen and digoxin, but not epoprostenol. Control was placebo plus current therapy.
The primary endpoint for each study was change in 6-minute walk distance at 12 weeks for the first study and 16 weeks for the second study. In both studies, treatment with bosentan resulted in significant increases in exercise capacity. The placebo-corrected increases in walk distance compared with baseline were 76 metres (p = 0.02; t-test) and 44 metres (p = 0.0002; Mann-Whitney U test) at the primary endpoint of each study, respectively. The differences between the two groups, 125 mg twice daily and 250 mg twice daily, were not statistically significant but there was a trend towards improved exercise capacity in the group treated with 250 mg twice daily.
The improvement in walk distance was apparent after 4 weeks of treatment, was clearly evident after 8 weeks of treatment and was maintained for up to 28 weeks of double-blind treatment in a subset of the patient population.
In a retrospective responder analysis based on change in walking distance, WHO functional class and dyspnoea of the 95 patients randomised to bosentan 125 mg twice daily in the placebo-controlled studies, it was found that at week 8, 66 patients had improved, 22 were stable and 7 had deteriorated. Of the 22 patients stable at week 8, 6 improved at week 12/16 and 4 deteriorated compared with baseline. Of the 7 patients who deteriorated at week 8, 3 improved at week 12/16 and 4 deteriorated compared with baseline.
Invasive haemodynamic parameters were assessed in the first study only. Treatment with bosentan led to a significant increase in cardiac index associated with a significant reduction in pulmonary artery pressure, pulmonary vascular resistance and mean right atrial pressure.
A reduction in symptoms of PAH was observed with bosentan treatment. Dyspnoea measurement during walk tests showed an improvement in bosentan-treated patients. In the AC-052-352 study, 92% of the 213 patients were classified at baseline as WHO functional class III and 8% as class IV. Treatment with bosentan led to a WHO functional class improvement in 42.4% of patients (placebo 30.4%). The overall change in WHO functional class during both studies was significantly better among bosentan-treated patients as compared with placebo-treated patients. Treatment with bosentan was associated with a significant reduction in the rate of clinical worsening compared with placebo at 28 weeks (10.7% vs 37.1%, respectively; p = 0.0015).
In a randomised, double-blind, multi-centre, placebo-controlled study (AC-052-364 [EARLY]), 185 PAH patients in WHO functional class II (mean baseline 6-minute walk distance of 435 metres) received bosentan 62.5 mg twice daily for 4 weeks followed by 125 mg twice daily (n = 93), or placebo (n = 92) for 6 months. Enrolled patients were PAH-treatment-naïve (n = 156) or on a stable dose of sildenafil (n = 29). The co-primary endpoints were percentage change from baseline in pulmonary vascular resistance (PVR) and change from baseline in 6-minute walk distance to Month 6 versus placebo. The table as follows illustrates the pre-specified protocol analyses. (See Table 1.)
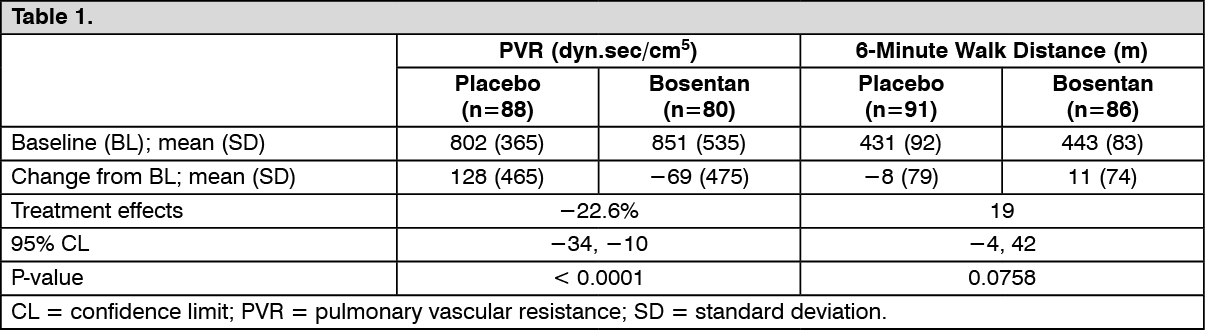 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment with bosentan was associated with a reduction in the rate of clinical worsening, defined as a composite of symptomatic progression, hospitalisation for PAH and death, compared with placebo (proportional risk reduction 77%, 95% confidence interval [CI] 20-94%, p = 0.0114). The treatment effect was driven by improvement in the component symptomatic progression. There was one hospitalisation related to PAH worsening in the bosentan group and three hospitalisations in the placebo group. Only one death occurred in each treatment group during the 6-month double-blind study period, therefore no conclusion can be drawn on survival.
Long-term data were generated from all 173 patients who were treated with bosentan in the controlled phase and/or were switched from placebo to bosentan in the open-label extension phase of the EARLY study. The mean duration of exposure to bosentan treatment was 3.6 ± 1.8 years (up to 6.1 years), with 73% of patients treated for at least 3 years and 62% for at least 4 years. Patients could receive additional PAH treatment as required in the open-label extension. The majority of patients were diagnosed with idiopathic or heritable PAH (61%). Overall, 78% of patients remained in WHO functional class II. Kaplan-Meier estimates of survival were 90% and 85% at 3 and 4 years after the start of treatment, respectively. At the same time points, 88% and 79% of patients remained free from PAH worsening (defined as all-cause death, lung transplantation, atrial septostomy or start of intravenous or subcutaneous prostanoid treatment). The relative contributions of previous placebo treatment in the double-blind phase and of other medications started during the open-label extension period are unknown.
In a prospective, multi-centre, randomised, double-blind, placebo-controlled study (AC-052-405 [BREATHE-5]), patients with PAH WHO functional class III and Eisenmenger physiology associated with congenital heart disease received bosentan 62.5 mg twice daily for 4 weeks, then 125 mg twice daily for a further 12 weeks (n = 37, of whom 31 had a predominantly right to left, bidirectional shunt). The primary objective was to show that bosentan did not worsen hypoxaemia. After 16 weeks, the mean oxygen saturation was increased in the bosentan group by 1.0% (95% CI -0.7%-2.8%) as compared to the placebo group (n = 17), showing that bosentan did not worsen hypoxaemia. The mean pulmonary vascular resistance was significantly reduced in the bosentan group (with a predominant effect observed in the subgroup of patients with bidirectional intracardiac shunt). After 16 weeks, the mean placebo-corrected increase in 6-minute walk distance was 53 metres (p = 0.0079), reflecting improvement in exercise capacity. Twenty-six patients continued to receive bosentan in the 24-week open-label extension phase (AC-052-409) of the BREATHE-5 study (mean duration of treatment = 24.4 ± 2.0 weeks) and, in general, efficacy was maintained.
An open-label, non-comparative study (AC-052-362 [BREATHE-4]) was performed in 16 patients with WHO functional class III PAH associated with HIV infection. Patients were treated with bosentan 62.5 mg twice daily for 4 weeks followed by 125 mg twice daily for a further 12 weeks. After 16 weeks' treatment, there were significant improvements from baseline in exercise capacity: the mean increase in 6-minute walk distance was 91.4 metres from 332.6 metres on average at baseline (p < 0.001). No formal conclusion can be drawn regarding the effects of bosentan on antiretroviral drug efficacy (see also Precautions).
There are no studies to demonstrate beneficial effects of Tracleer treatment on survival. However, long-term vital status was recorded for all 235 patients who were treated with bosentan in the two pivotal placebo-controlled studies (AC-052-351 and AC-052-352) and/or their two uncontrolled, open-label extensions. The mean duration of exposure to bosentan was 1.9 years ± 0.7 years (min 0.1 years; max: 3.3 years) and patients were observed for a mean of 2.0 ± 0.6 years. The majority of patients were diagnosed as primary pulmonary hypertension (72%) and were in WHO functional class III (84%). In this total population, Kaplan-Meier estimates of survival were 93% and 84% 1 and 2 years after the start of treatment with bosentan, respectively. Survival estimates were lower in the subgroup of patients with PAH secondary to systemic sclerosis. The estimates may have been influenced by the initiation of epoprostenol treatment in 43/235 patients.
Combination with epoprostenol: The combination of bosentan and epoprostenol has been investigated in two studies: AC-052-355 (BREATHE-2) and AC-052-356 (BREATHE-3). AC-052-355 was a multi-centre, randomised, double-blind, parallel-group study of bosentan versus placebo in 33 patients with severe PAH who were receiving concomitant epoprostenol therapy. AC-052-356 was an open-label, uncontrolled study; 10 of the 19 paediatric patients were on concomitant bosentan and epoprostenol therapy during the 12-week study. The safety profile of the combination was not different from the one expected with each component and the combination therapy was well tolerated in adults. The clinical benefit of the combination has not been demonstrated.
Pharmacokinetics: The pharmacokinetics of bosentan have mainly been documented in healthy subjects. Limited data in patients show that the exposure to bosentan in adult PAH patients is approximately 2-fold greater than in healthy adult subjects.
In healthy subjects, bosentan displays dose- and time-dependent pharmacokinetics. Clearance and volume of distribution decrease with increased intravenous doses and increase with time. After oral administration, the systemic exposure is proportional to dose up to 500 mg. At higher oral doses, Cmax and AUC increase less than proportionally to the dose.
Absorption: In healthy subjects, the absolute bioavailability of bosentan is approximately 50% and is not affected by food. The maximum plasma concentrations are attained within 3-5 hours.
Distribution: Bosentan is highly bound (> 98%) to plasma proteins, mainly albumin. Bosentan does not penetrate into erythrocytes.
A volume of distribution (Vss) of about 18 litres was determined after an intravenous dose of 250 mg.
Biotransformation and elimination: After a single intravenous dose of 250 mg, the clearance was 8.2 L/h. The terminal elimination half-life (t½) is 5.4 hours.
Upon multiple dosing, plasma concentrations of bosentan decrease gradually to 50-65% of those seen after single dose administration. This decrease is probably due to auto-induction of metabolising liver enzymes. Steady-state conditions are reached within 3-5 days.
Bosentan is eliminated by biliary excretion following metabolism in the liver by the cytochrome P450 isoenzymes, CYP2C9 and CYP3A4. Less than 3% of an administered oral dose is recovered in urine.
Bosentan forms three metabolites and only one of these is pharmacologically active. This metabolite is mainly excreted unchanged via the bile. In adult patients, the exposure to the active metabolite is greater than in healthy subjects. In patients with evidence of the presence of cholestasis, the exposure to the active metabolite may be increased.
Bosentan is an inducer of CYP2C9 and CYP3A4 and possibly also of CYP2C19 and the P-glycoprotein. In vitro, bosentan inhibits the bile salt export pump in hepatocyte cultures.
In vitro data demonstrated that bosentan had no relevant inhibitory effect on the CYP isoenzymes tested (CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 2E1, 3A4). Consequently, bosentan is not expected to increase the plasma concentrations of medicinal products metabolised by these isoenzymes.
Pharmacokinetics in special populations: Based on the investigated range of each variable, it is not expected that the pharmacokinetics of bosentan will be influenced by gender, body weight, race, or age in the adult population to any relevant extent.
Hepatic impairment: In patients with mildly impaired liver function (Child-Pugh class A) no relevant changes in the pharmacokinetics have been observed. The steady-state AUC of bosentan was 9% higher and the AUC of the active metabolite, Ro 48-5033, was 33% higher in patients with mild hepatic impairment than in healthy volunteers.
The impact of moderately impaired liver function (Child-Pugh class B) on the pharmacokinetics of bosentan and its primary metabolite Ro 48-5033 was investigated in a study including 5 patients with pulmonary hypertension associated with portal hypertension and Child-Pugh class B hepatic impairment, and 3 patients with PAH from other causes and normal liver function. In the patients with Child-Pugh class B liver impairment, the mean (95% CI) steady-state AUC of bosentan was 360 (212=613) ng·h/mL, i.e., 4.7 times higher, and the mean (95% CI) AUC of the active metabolite Ro 48-5033 was 106 (58.4-192) ng⋅h/mL, i.e., 12.4 times higher than in the patients with normal liver function (bosentan: mean [95% CI] AUC: 76.1 [9.07-638] ng⋅h/mL; Ro 48-5033: mean [95% CI] AUC 8.57 [1.28-57.2] ng·h/mL). Though the number of patients included was limited and with high variability, these data indicate a marked increase in the exposure to bosentan and its primary metabolite Ro 48-5033 in patients with moderate liver function impairment (Child-Pugh class B).
The pharmacokinetics of bosentan have not been studied in patients with Child-Pugh class C hepatic impairment. Tracleer is contraindicated in patients with moderate to severe hepatic impairment, i.e., Child-Pugh class B or C (see Contraindications).
Renal impairment: In patients with severe renal impairment (creatinine clearance 15-30 mL/min), plasma concentrations of bosentan decreased by approximately 10%. Plasma concentrations of bosentan metabolites increased about 2-fold in these patients as compared with subjects with normal renal function. No dose adjustment is required in patients with renal impairment. There is no specific clinical experience in patients undergoing dialysis. Based on physicochemical properties and the high degree of protein binding, bosentan is not expected to be removed from the circulation by dialysis to any significant extent (see Dosage & Administration).
Toxicology: Preclinical safety data: A 2-year carcinogenicity study in mice showed an increased combined incidence of hepatocellular adenomas and carcinomas in males, but not in females, at plasma concentrations about 2 to 4 times the plasma concentrations achieved at the therapeutic dose in humans. In rats, oral administration of bosentan for 2 years produced a small, significant increase in the combined incidence of thyroid follicular cell adenomas and carcinomas in males, but not in females, at plasma concentrations about 9 to 14 times the plasma concentrations achieved at the therapeutic dose in humans. Bosentan was negative in tests for genotoxicity. There was evidence of a mild thyroid hormonal imbalance induced by bosentan in rats. However, there was no evidence of bosentan affecting thyroid function (thyroxine, TSH) in humans.
The effect of bosentan on mitochondrial function is unknown.
Bosentan has been shown to be teratogenic in rats at plasma levels higher than 1.5 times the plasma concentrations achieved at the therapeutic dose in humans. Teratogenic effects, including malformations of the head and face and of the major vessels, were dose dependent. The similarities of the pattern of malformations observed with other ET receptor antagonists and in ET knock-out mice indicate a class effect. Appropriate precautions must be taken for women of childbearing potential (see Contraindications, Precautions and Use in Pregnancy & Lactation).
Development of testicular tubular atrophy and impaired fertility has been linked with chronic administration of endothelin receptor antagonists in rodents.
In fertility studies in male and female rats, no effects on sperm count, motility and viability, or on mating performance or fertility were observed at exposures that were 21 and 43 times the expected therapeutic level in humans, respectively; nor was there any adverse effect on the development of the pre-implantation embryo or on implantation.
Slightly increased incidence of testicular tubular atrophy was observed in rats given bosentan orally at doses as low as 125 mg/kg/day (about 4 times the maximum recommended human dose [MRHD] and the lowest doses tested) for two years but not at doses as high as 1 500 mg/kg/day (about 50 times the MRHD) for 6 months. In a juvenile rat toxicity study, where rats were treated from Day 4 post partum up to adulthood, decreased absolute weights of testes and epididymides, and reduced number of sperm in epididymides were observed after weaning. The NOAEL was 21 times (at Day 21 post partum) and 2.3 times (Day 69 post partum) the human therapeutic exposure, respectively.
However, no effects on general development, growth, sensory, cognitive function and reproductive performance were detected at 7 (males) and 19 (females) times the human therapeutic exposure at Day 21 post partum. At adult age (Day 69 post partum), no effects of bosentan were detected at 1.3 (males) and 2.6 (females) times the therapeutic exposure in children with PAH.