The information highlighted (if any) are the most recent updates for this brand.
Dexketoprofen, tramadol hydrochloride.
Each tablet contains: 75 mg of tramadol hydrochlorideand 25 mg of dexketoprofen.
The score line is only to facilitate breaking for ease of swallowing and not to divide into equal doses.
Excipients with known effects: each tablet contains 33.07 mg croscarmellose sodium and 1.83 mg sodium stearyl fumarate.
Excipients/Inactive Ingredients: Tablet Core: Microcrystalline cellulose; Maize Starch, pregelatinised; Croscarmellose sodium; Sodium stearyl fumarate; Anhydrous colloidal silica.
Film-coating: Polyvinyl alcohol; Titanium dioxide; Macrogol/PEG 3350; Talc.
Pharmacotherapeutic group: Opioids in combination with non-opioid analgesics. ATC code: N02AJ14.
Pharmacology: Pharmacodynamics: Mechanism of action: Dexketoprofen is the tromethamine salt of S-(+)-2-(3-benzoylphenyl)propionic acid, an analgesic, anti-inflammatory and antipyretic drug, which belongs to the non-steroidal anti-inflammatory group of drugs (M01AE).
The mechanism of action of non-steroidal anti-inflammatory drugs is related to the reduction of prostaglandin synthesis by the inhibition of cyclooxygenase pathway. Specifically, there is an inhibition of the transformation of arachidonic acid into cyclic endoperoxides, PGG2 and PGH2, which produce prostaglandins PGE1, PGE2, PGF2α and PGD2 and also prostacyclin PGI2 and thromboxanes (TxA2 and TxB2). Furthermore, the inhibition of the synthesis of prostaglandins could affect other inflammation mediators such as kinins, causing an indirect action which would be additional to the direct action.
Dexketoprofen has been demonstrated to be an inhibitor for COX-1 and COX-2 activities in experimental animals and humans.
Tramadol hydrochloride is a centrally acting synthetic opioid analgesic. It is a non-selective, partial agonist of μ-, δ- and κ-opioid receptors with a higher affinity for μ-receptors. Opioid activity is due to both low affinity binding of the parent compound and higher affinity binding of the O-demethylated metabolite M1 to μ-opioid receptors. In animal models, M1 is up to 6 times more potent than tramadol in producing analgesia and 200 times more potent in μ-opioid binding. Tramadol-induced analgesia is only partially antagonized by the opiate antagonist naloxone in several animal tests. The relative contribution of both tramadol and M1 to human analgesia is dependent upon the plasma concentrations of each compound.
Tramadol has been shown to inhibit reuptake of norepinephrine and serotonin in vitro, as have some other opioid analgesics. These mechanisms may contribute independently to the overall analgesic profile of tramadol.
Tramadol has an antitussive action. In contrast to morphine, analgesic doses of tramadol over a wide range have no respiratory depressant effect. Also gastrointestinal motility is less affected. Effects on the cardiovascular system tend to be slight. The potency of tramadol is reported to be 1/10 (one tenth) to 1/6 (one sixth) that of morphine.
Pharmacodynamic effects: Preclinical studies have shown a synergistic interaction between the active ingredients observed during both acute and chronic inflammation models and suggest that lower doses of each active ingredient allow to obtain effective analgesia.
Clinical efficacy and safety: Clinical studies performed on several models of moderate to severe nociceptive pain (including dental pain, somatic pain and visceral pain) demonstrated effective analgesic activity of Skudexa.
In a multiple-dose, double-blind, randomised, parallel group study in 606 patients with moderate to severe pain after abdominal hysterectomy, mean age 47.6 (range 25 to 73), the analgesic efficacy of the combination versus the individual components was assessed by means of the sum of pain intensity difference values over the interval of 8 hours (SPID8) after the first dose of study medication, with pain intensity been assessed on a 100mm visual analogue scale (VAS). Higher value of SPID indicates greater pain relief. The treatment with Skudexa resulted in an analgesic effect significantly greater than those of the individual components given at the same dose (dexketoprofen 25 mg) or at a higher dose (tramadol 100mg), being the results as follows: Skudexa (241.8), dexketoprofen 25 mg (184.5), tramadol 100 mg (157.3).
Over the first 8 hours following Skudexa, patients reported a significantly lower Pain Intensity (mean PI-VAS= 33.6) with a statistically significant (p< 0.0001) difference over dexketoprofen 25 mg (mean PI-VAS= 42.6) and tramadol 100 mg (mean PI-VAS= 42.9). Superior analgesia was also demonstrated over 56 hours following repeated doses administered according to the posology scheme in an ITT population in which patients who did not receive active treatment as first single dose were excluded, with statistically significant (p< 0.0001) difference between Skudexa and dexketoprofen 25 mg (-8.4) and tramadol 100 mg (-5.5).
Patients treated with Skudexa were in need of less rescue medication to control pain (11.8% of patients in comparison with 21.3% (p= 0.0104) and 21.4% (p= 0.0097) under dexketoprofen 25 mg and tramadol 100 mg, respectively). When the impact of rescue medication use is taken into account, the superior analgesic effect of Skudexa in the repeat use over 56 hours becomes more evident, reaching a difference in PI-VAS favouring Skudexa over dexketoprofen (-11.0) and tramadol (-9.1) with a statistical significance of p= <0.0001.
In a multiple-dose, double-blind, randomised, parallel group study in 641 patients with moderate to severe pain after total hip arthroplasty, mean age 61.9 (range 29 to 80), the analgesic efficacy of the combination versus the individual components was assessed over 8 hours after the first dose of study medication (SPID8). The treatment with Skudexa resulted in an analgesic effect significantly greater than those of the individual components given at the same dose (dexketoprofen 25mg) or at a higher dose (tramadol 100mg); Skudexa (246.9), dexketoprofen 25 mg (208.8), tramadol 100 mg (204.6). Over the first 8 hours following Skudexa, patients reported a significantly lower Pain Intensity (mean PI-VAS= 26.3) with a statistically significant (p< 0.0001) difference over dexketoprofen 25 mg (mean PI-VAS= 33.6) and tramadol 100 mg (mean PI-VAS= 33.7).
Superior analgesia was also demonstrated over 56 hours following repeated doses administered according to the posology scheme in an ITT population in which patients who did not receive active treatment as first single dose were excluded, with statistically significant (p< 0.0001) difference between Skudexa and dexketoprofen 25 mg (-8.1) and tramadol 100 mg (-6.3), respectively.
Rescue medication to control pain was required by 15.5% of patients under Skudexa, in comparison with 28.0% (p= 0.0017) and 25.2% (p=0.0125) under dexketoprofen 25 mg and tramadol 100 mg, respectively. When the impact of rescue medication use is taken into account, the superior analgesic effect of Skudexa in the repeat use over 56 hours becomes more evident, reaching a statistical (p= <0.0001) difference in PI-VAS favouring Skudexa over dexketoprofen (-10.4) and tramadol (-8.3).
Paediatric population: The European Medicines Agency has waived the obligation to submit the results of studies with Skudexa in all subsets of the paediatric population in the treatment of moderate to severe acute pain (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Concomitant administration of dexketoprofen and tramadol had no effects on the pharmacokinetic parameters of either component in healthy subjects.
In normal healthy adults, peak plasma concentrations of dexketoprofen and tramadol are reached in about 30 min (range 15 to 60 min) and 1.6 to 2 hours, respectively.
Dexketoprofen: Absorption: After oral administration of dexketoprofen to humans, the Cmax is reached at 30 min (range 15 to 60 min).
When administered concomitantly with food, the AUC does not change, however the Cmax of dexketoprofen decreases and its absorption rate is delayed (increased tmax).
Distribution: The distribution half-life and elimination half-life values of dexketoprofen are 0.35 and 1.65 hours, respectively. As with other drugs with a high plasma protein binding (99%), its volume of distribution has a mean value below 0.25 l/kg.
In multiple-dose pharmacokinetic studies, it was observed that the AUC after the last administration is not different from that obtained following a single dose, indicating that no drug accumulation occurs.
Biotransformation and Elimination: After administration of dexketoprofen only the S-(+) enantiomer is obtained in urine, demonstrating that no conversion to the R-(-) enantiomer occurs in humans.
The main elimination route for dexketoprofen is glucuronide conjugation followed by renal excretion.
Tramadol: Absorption: More than 90% of tramadol is absorbed after oral administration. The mean absolute bioavailability is approximately 70%, irrespective of concomitant intake of food.
The difference between absorbed and non-metabolised available tramadol is probably due to low first-pass effect. The first-pass effect after oral administration is a maximum of 30%. Tramadol has a high tissue affinity (Vd,β=203 ± 40 l). Protein binding is about 20%.
Following a single oral dose administration of tramadol 100 mg as capsules or tablets to young healthy volunteers, plasma concentrations were detectable within approximately 15 to 45 minutes within a mean Cmax of 280 to 208 mcg/L and Tmax of 1.6 to 2h.
Distribution: Tramadol passes the blood-brain and placenta barrier. Very small amounts of the substance and its O-desmethyl derivative are found in the breast milk (0.1% and 0.02% respectively of the applied dose).
Biotransformation: In humans tramadol is mainly metabolised by means of N- and O-demethylation and conjugation of the O-demethylation products with glucuronic acid. Only O-desmethyltramadol is pharmacologically active. There are considerable interindividual quantitative differences between the other metabolites. So far, eleven metabolites have been found in the urine. Animal experiments have shown that O-desmethyltramadol is more potent than the parent substance by the factor 2 - 4. Its half life t½β (6 healthy volunteers) is 7.9 h (range 5.4 - 9.6 h) and is approximately that of tramadol.
The inhibition of one or both cytochrome P450 isoenzymes, CYP3A4 and CYP2D6 involved in the metabolism of tramadol, may affect the plasma concentration of tramadol or its active metabolite.
Elimination: Elimination half-life t½β is approximately 6 h, irrespective of the mode of administration. In patients above 75 years of age it may be prolonged by a factor of approximately 1.4.
Tramadol and its metabolites are almost completely excreted via the kidneys. Cumulative urinary excretion is 90% of the total radioactivity of the administered dose. In cases of impaired hepatic and renal function the half-life may be slightly prolonged. In patients with cirrhosis of the liver, elimination half-lives of 13.3 ± 4.9 h (tramadol) and 18.5 ± 9.4 h (O-desmethyltramadol), in an extreme case 22.3 h and 36 h respectively have been determined. In patients with renal insufficiency (creatinine clearance < 5 ml/min) the values were 11 ± 3.2 h and 16.9 ± 3 h, in an extreme case 19.5 h and 43.2 h, respectively.
Linearity/non-linearity: Tramadol has a linear pharmacokinetic profile within the therapeutic dosage range.
The relationship between serum concentrations and the analgesic effect is dose-dependent, but varies considerably in isolated cases. A serum concentration of 100 - 300 ng/ml is usually effective.
Toxicology: Preclinical safety data: Tramadol hydrochloride-dexketoprofen combination: Preclinical data with the combination revealed no special hazard for humans based on conventional studies of safety pharmacology and repeated dose toxicity.
The combination of dexketoprofen and tramadol had not significant effect on cardiovascular system as assessed by both in vitro and in vivo tests. Less effect on gastrointestinal transit were observed with the combination as compared to tramadol alone.
A 13-week chronic toxicity study in rats, gave No Observed Adverse Effect Levels (NOAELs) of 6 mg/kg/day for dexketoprofen and 36 mg/kg/day for tramadol (highest tested doses), when administered both singularly or in combination (corresponding to AUC-based exposures at the NOAEL after single doses of 25.10 times and 1.38 times the human exposure to dexketoprofen and tramadol, respectively, at a single clinical dose of 25 mg dexketoprofen and 75 mg tramadol).
No new toxicities, different from those previously described for dexketoprofen or tramadol were observed.
Dexketoprofen: Preclinical data on dexketoprofen revealed no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, toxicity to reproduction and immunopharmacology. The chronic toxicity studies carried out in mice and monkeys gave a No Observed Adverse Effect Level (NOAEL) of 3 mg/kg/day. The main adverse effect observed at high doses was gastrointestinal erosions and ulcers that developed dose-dependently.
Tramadol: In repeated oral and parenteral administration of tramadol during 6 to 26 weeks to rats and dogs and oral administration for 12 months in dogs haematological, clinico-chemical and histological investigations showed no evidence of any substance-related changes. Central nervous manifestations only occurred after high doses considerably above the therapeutic range: restlessness, salivation, convulsions, and reduced weight gain. Rats and dogs tolerated oral doses of 20 mg/kg and 10 mg/kg body weight respectively, and dogs rectal doses of 20 mg/kg body weight without any reactions.
In rats, tramadol dosages from 50 mg/kg/day upwards caused toxic effects in dams and raised neonate mortality. In the offspring retardation occurred in the form of ossification disorders and delayed vaginal and eye opening. Male fertility was not affected. After higher doses (from 50 mg/kg/day upwards) females exhibited a reduced pregnancy rate. In rabbits there were toxic effects in dams from 125 mg/kg upwards and skeletal anomalies in the offspring.
In some in-vitro test systems there was evidence of mutagenic effects. In-vivo studies showed no such effects.
According to knowledge gained so far, tramadol can be classified as non-mutagenic.
Studies on the tumorigenic potential of tramadol hydrochloride have been carried out in rats and mice. The study in rats showed no evidence of any substance-related increase in the incidence of tumours. In the study in mice there was an increased incidence of liver cell adenomas in male animals (a dose-dependent, non-significant increase from 15 mg/kg upwards) and an increase in pulmonary tumours in females of all dosage groups (significant, but not dose-dependent).
Symptomatic short term treatment of moderate to severe acute pain in adult patients whose pain is considered to require a combination of tramadol and dexketoprofen.
Posology: Adults: The recommended dosage is one film-coated tablet (corresponding to 75 mg of tramadol hydrochloride and 25 mg of dexketoprofen). Additional doses can be taken as needed, with a minimum dosing interval of 8 hours. The total daily dose should not exceed three film-coated tablets per day (corresponding to 225 mg of tramadol hydrochloride and 75 mg of dexketoprofen).
Skudexa is intended for short term use only and the treatment must be strictly limited to the symptomatic period and in any case not more than 5 days.
Switching to a single agent analgesia should be considered according to pain intensity and response of the patient.
Undesirable effects may be minimised by using the lowest number of doses for the shortest duration necessary to control symptoms. (See Precautions).
Elderly: In elderly patients the starting recommended dosage is one film-coated tablet; additional doses can be taken as needed with the minimum dose interval of 8 hours and not exceeding the total daily dose of 2 film-coated tablets (corresponding to 150 mg of tramadol hydrochloride and 50 mg of dexketoprofen). The dosage may be increased to a maximum of 3 daily film-coated tablets as recommended for the general population only after good general tolerance has been ascertained.
Limited data are available in patients over 75 years, therefore SKUDEXA should be used with caution in these patients (see Precautions).
Hepatic impairment: Patients with mild to moderate hepatic dysfunction should start therapy at reduced number of doses (total daily dose 2 film-coated tablets Skudexa) and be closely monitored.
Skudexa should not be used in patients with severe hepatic dysfunction (see Contraindications).
Renal impairment: The initial total daily dosage should be reduced to 2 film-coated tablets Skudexa in patients with mildly impaired renal function (creatinine clearance 60 - 89 ml/min) (see Precautions).
Skudexa should not be used in patients with moderate to severe renal dysfunction (creatinine clearance ≤59 ml/min) (see Contraindications).
Paediatric population: The safety and efficacy of Skudexa has not been studied in paediatric population. Therefore Skudexa should not be used in children and adolescents.
Method of administration: Oral use.
Skudexa should be swallowed with a sufficient amount of fluid (e.g. one glass of water). Concomitant administration with food delays the absorption rate of the drug (see Pharmacology: Pharmacokinetics under Actions), for a faster effect the tablets may be taken at least 30 minutes before meals.
No cases of overdose have been reported in the clinical studies. Data reported for dexketoprofen and tramadol as single agents should be taken into account.
Symptoms: Dexketoprofen: The symptomatology following overdose due to dexketoprofen is not known.
Medicinal products containing dexketoprofen have produced gastrointestinal (vomiting, anorexia, abdominal pain) and neurological (somnolence, vertigo, disorientation, headache) disorders.
Tramadol: In tramadol overdose, in principle, the same symptoms occur as for all other central acting analgesics (opioids). In particular, these include miosis, vomiting, cardiovascular collapse, consciousness disorders up to coma, convulsions and respiratory depression up to respiratory arrest.
Management: Dexketoprofen: In case of accidental or excessive intake, immediately initiate symptomatic therapy according to the patient's clinical condition.
If more than 5 mg/kg has been ingested by an adult or a child, activated charcoal should be administered within the first hour after ingestion. Dexketoprofen may be removed by dialysis.
Tramadol: Keep the respiratory tract open (and avoid aspiration), maintain respiration and circulation depending on the symptoms. The antidote for respiratory depression is naloxone. In animal experiments naloxone had no effect on convulsions. In such case diazepam should be given intravenously.
In case of orally intoxication, gastrointestinal decontamination with activated charcoal is recommended within two hours after tramadol intake.
Tramadol may be removed by dialysis, but it is minimally eliminated from the serum by haemodialysis or haemofiltration. Therefore treatment of acute intoxication with tramadol with haemodialysis or haemofiltration alone is not suitable for detoxification.
The contraindications reported for dexketoprofen and tramadol as single agents should be taken into account.
Dexketoprofen must not be administered in the following cases: hypersensitivity to dexketoprofen, to any other NSAID, or to any of the excipients listed in Description; patients in whom substances with a similar action (e.g. acetylsalicylic acid, or other NSAIDs) precipitate attacks of asthma, bronchospasm, acute rhinitis, or cause nasal polyps, urticaria or angioneurotic oedema; known photoallergic or phototoxic reactions during treatment with ketoprofen or fibrates; patients with active peptic ulcer/gastrointestinal haemorrhage or any history of gastrointestinal bleeding ulceration or perforation; patients with history of gastrointestinal bleeding or perforation, related to previous NSAIDs therapy; patiens with chronic dyspepsia; patients who have other active bleedings or bleeding disorders; patients with Crohn's disease or ulcerative colitis; patients with severe heart failure; patients with moderate to severe renal dysfunction (creatinine clearance ≤59 ml/min); patients with severely impaired hepatic function (Child-Pugh C); patients with haemorrhagic diathesis and other coagulation disorders; patients with severe dehydration (caused by vomiting, diarrhoea or insufficient fluid intake).
Tramadol must not be administered in the following cases: hypersensitivity to tramadol or to any of the excipients listed in Description; in acute intoxication with alcohol, hypnotics, analgesics, opioids or psychotropic medicinal products; in patients receiving MAO inhibitors, or who have taken them within the last 14 days (see Interactions); in patients with epilepsy not adequately controlled by treatment (see Precautions); severe respiratory depression; Children younger than 18 years to treat pain after surgery to remove the tonsils and or adenoids; Adolescence between 12 and 18 years who are obese or have conditions such as obstructive sleep apnea or severe lung disease, which may increase the risk of serious breathing problems.
Skudexa is contraindicated during pregnancy and lactation (see Use in Pregnancy & Lactation).
The special warnings and precautions reported for dexketoprofen and tramadol as single agents should be taken into account.
Dexketoprofen: Administer with caution in patients with a history of allergic conditions.
The use of dexketoprofen with concomitant other NSAIDs including cyclooxygenase-2 selective inhibitors should be avoided (see Interactions).
Undesirable effects may be minimised by using the lowest effective dose for the shortest duration necessary to control symptoms (see Dosage & Administration, and gastrointestinal GI and cardiovascular risks as follows).
Gastrointestinal safety: Gastrointestinal bleeding, ulceration or perforation which can be fatal, have been reported with all NSAIDs at anytime during treatment, with or without warning symptoms or a previous history of serious gastrointestinal events. When gastrointestinal bleeding or ulceration occurs in patients receiving dexketoprofen, the treatment should be withdrawn.
The risk of gastrointestinal bleeding, ulceration or perforation is higher with increasing NSAID doses, in patients with a history of ulcer, particularly if complicated with haemorrhage or perforation (see Contraindications), and in older people.
As with all NSAIDs, any history of oesophagitis, gastritis and/or peptic ulcer must be identified in order to ensure their total cure before starting treatment with dexketoprofen. Patients with gastrointestinal symptoms or history of gastrointestinal disease should be monitored for digestive disturbances, especially gastrointestinal bleeding.
NSAIDs should be used with caution in patients with a history of gastrointestinal disease (ulcerative colitis, Crohn's disease) as their condition may be exacerbated (see Adverse Reactions).
Combination therapy with protective agents (e.g. misoprostol or proton pump inhibitors) should be considered for these patients, and also for patients requiring concomitant low dose acetylsalicylic acid, or other drugs likely to increase gastrointestinal risk (see as follows and Interactions).
Patients with a history of gastrointestinal toxicity, particularly when elderly, should report any unusual abdominal symptoms (especially gastrointestinal bleeding) particularly in the initial stages of treatment.
Caution should be advised in patients receiving concomitant medications which could increase the risk of ulceration or bleeding, such as oral corticosteroids, anticoagulants such as warfarin, selective serotonin-reuptake inhibitors or anti-platelet agents such as acetylsalicylic acid (see Interactions).
Renal safety: Caution should be exercised in patients with impairment of renal functions. In these patients, the use of NSAIDs may result in deterioration of renal function, fluid retention and oedema. Caution is also required in patients receiving diuretic therapy or those who could develop hypovolaemia as there is an increased risk of nephrotoxicity.
Adequate fluid intake should be ensured during treatment to prevent dehydration and possibly associated increased renal toxicity.
As with all NSAIDs, it can increase plasma urea nitrogen and creatinine. As with other inhibitors of prostaglandin synthesis, it can be associated with adverse effects on the renal system which can lead to glomerular nephritis, interstitial nephritis, renal papillary necrosis, nephrotic syndrome and acute renal failure.
Liver safety: Caution should be exercised in patients with impairment of hepatic functions. As with other NSAIDs, it can cause transient small increases in some liver parameters, and also significant increases in aspartate transaminase (AST) also known as serum glutamic oxaloacetic transaminase (SGOT) and Alanine transaminase (ALT), also known as serum glutamic-pyruvic transaminase (SGPT). In case of a relevant increase in such parameters, therapy must be discontinued.
Cardiovascular and cerebrovascular safety: Appropriate monitoring and advice are required for patients with a history of hypertension and/or mild to moderate congestive heart failure as fluid retention and oedema have been reported in association with NSAIDs therapy. Special caution should be exercised in patients with a history of cardiac disease, in particular those with previous episodes of heart failure as there is an increased risk of triggering heart failure.
Clinical trial and epidemiological data suggest that use of some NSAIDs (particularly at high doses and in long term treatment) may be associated with a small increase in the risk of arterial thrombotic events (for example myocardial infarction or stroke). There are insufficient data to exclude such a risk for dexketoprofen.
Patients with uncontrolled hypertension, congestive heart failure, established ischaemic heart disease, peripheral arterial disease, and/or cerebrovascular disease should only be treated with dexketoprofen after careful consideration. Similar consideration should be made before initiating long-term treatment of the patients with risk factors for cardiovascular disease (e.g. hypertension, hyperlipidaemia, diabetes mellitus, smoking).
All non-selective NSAIDs can inhibit platelet aggregation and prolong bleeding time via inhibition of prostaglandin synthesis. Therefore, the use of dexketoprofen in patients who are receiving other therapy that interferes with haemostasis, such as warfarin or other coumarins or heparins is not recommended (see Interactions).
Skin reactions: Serious skin reactions, some of them fatal, including exfoliative dermatitis, Stevens-Johnson syndrome, and toxic epidermal necrolysis, have been reported very rarely in association with the use of NSAIDs (see Adverse Reactions). Patients appear to be at highest risk of these reactions early in the course of therapy, the onset of the reaction occurring in the majority of cases within the first month of treatment. Dexketoprofen should be discontinued at the first appearance of skin rash, mucosal lesions, or any other sign of hypersensitivity.
Other information: Particular caution is required in patients with: congenital disorder of porphyrin metabolism (e.g. acute intermittent porphyria); dehydration; directly after major surgery.
Severe acute hypersensitivity reactions (anaphylactic shock, for example) have been observed on very rare occasions. Treatment must be discontinued at the first signs of severe hypersensitivity reactions following intake of dexketoprofen. Depending on the symptoms, any medically required procedures must be initiated by specialist healthcare professionals.
Patients with asthma combined with chronic rhinitis, chronic sinusitis, and/or nasal polyposis have a higher risk of allergy to acetylsalicylic acid and/or NSAIDs than the rest of the population. Administration of this medicinal product can cause asthma attacks or bronchospasm, particularly in subjects allergic to acetylsalicylic acid or NSAIDs (see Contraindications).
Exceptionally, varicella can be at the origin of serious cutaneous and soft tissues infectious complications. To date, the contributing role of NSAIDs in the worsening of these infections cannot be ruled out. Thus, it is advisable to avoid use of dexketoprofen in case of varicella.
Dexketoprofen should be administered with caution to patients suffering from haematopoietic disorders, systemic lupus erythematosus or mixed connective tissue disease.
As other NSAIDs, dexketoprofen can mask the symptoms of infectious diseases.
Tramadol: Tramadol should be used with particular caution in addicted patients, patients with head injury, shock, a reduced level of consciousness of uncertain origin, disorders of the respiratory centre or function, or increased intracranial pressure.
In patients sensitive to opiates the product should be used with caution.
Care should be taken when treating patients with respiratory depression, or if concomitant CNS depressant drugs are being administered (see Interactions), or if the recommended dosage is significantly exceeded (see Overdosage) as the possibility of respiratory depression cannot be excluded in these situations.
Convulsions have been reported in patients receiving tramadol at the recommended dose levels. The risk may be increased when doses of tramadol exceed the recommended upper daily dose limit (400 mg).
In addition tramadol may increase the seizure risk in patients taking other medicinal products that lower the seizure threshold (see Interactions). Patients with epilepsy or those susceptible to seizures should only be treated with tramadol if there are compelling circumstances.
Tolerance, psychic and physical addiction may develop, especially after long-term use. In patients with a tendency to drug abuse or dependence, treatment with tramadol should only be carried out for short periods under strict medical supervision. When a patient no longer requires therapy with tramadol, it may be advisable to taper the dose gradually to prevent symptoms of withdrawal.
Risk from concomitant use of sedative medicines such as benzodiazepines or related drugs: Concomitant use of Skudexa and sedative medicines such as benzodiazepines or related drugs may result in sedation, respiratory depression, coma and death. Because of these risks, concomitant prescribing with these sedative medicines should be reserved for patients for whom alternative treatment options are not possible. If a decision is made to prescribe Skudexa concomitantly with sedative medicines, the lowest effective dose should be used, and the duration of treatment should be as short as possible.
The patients should be followed closely for signs and symptoms of respiratory depression and sedation. In this respect, it is strongly recommended to inform patients and their caregivers to be aware of these symptoms (see Interactions).
Respiratory depression: Administer Skudexa cautiously in patients at risk for respiratory depression, including patients with subtantially decreased respiratory reserve, hypoxia, hypercapnia, or pre-existing respiratory depression, as in these patients, even therapeutic doses of skudexa may decrease respiratory drive to point of apnea. In these patients, alternatively non-oploid analgesic should be considered. When large doses of tramadol are administrated with anaesthetic medications or alcohol respiratory depression may result. Respiratory depression should be treated as an overdose. If naloxone is to be administrated, use cautiously because it may precipitated seizures.
Cytochrome P450 (CYP) 2D6 Ultra-Rapid metabolism: Some individuals may be CYP2D6 ultra-rapid metabolisers. These individuals converts tramadol more rapidly than other people into its more potent oploid metabolites O-desmethyltramadol (M1). This rapid conversion could result in higher than expected oploid-like side effects including life-threathening respiratory depression. The prevalence of this CYP2D6 phenotype varies widely and has been estimatedly at 0.5 to 1% in Chinese, Japanese and Hispanics, 1 to 10% in Caucasians, 3% in African Americans, and 16-28% in North Africans, Ethiopians and Arabs. Data are not available for other ethnic groups.
Effects on ability to drive and use machines: The effects known for the single components of SKUDEXA apply to the fixed combination.
Dexketoprofen: Dexketoprofen has minor or moderate influence on the ability to drive and use machines, due to possible occurrence of dizziness or somnolence.
Tramadol: Even when taken according to instructions, tramadol may cause effects such as somnolence and dizziness and therefore may impair the reactions of drivers and machine operators. This applies particularly in conjunction with other psychotropic substances and alcohol.
Use
in the Elderly:Dexketoprofen: The elderly have an increased frequency of adverse reactions to NSAIDs especially gastrointestinal bleeding and perforation which may be fatal (see Dosage & Administration). These patients should commence treatment on the lowest dose available.
Elderly are more likely to be suffering from impaired renal cardiovascular or hepatic function (see Dosage & Administration).
Use in Children: Dexketoprofen: The safety and efficacy of Skudexa in children and adolescents have not been established. Therefore Skudexa should not be used in children and adolescents.
Tramadol: Post-operative use in children: There have been reports in the published literature that tramadol given post-operatively in children after tonsillectomy and/or adenoidectomy for obstructive sleep apnoea, led to rare, but life threatening adverse events. Extreme caution should be exercised when tramadol is administered to children for post-operative pain relief and should be accompanied by close monitoring for symptoms of opioid toxicity including respiratory depression.
Children with compromised respiratory function: Tramadol is not recommended for use in children in whom respiratory function might be compromised including neuromuscular disorders, severe cardiac or respiratory conditions, upper respiratory or lung infections, multiple trauma or extensive surgical procedures. These factors may worsen symptoms of opioid toxicity.
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, i.e. essentially "sodium-free".
Pregnancy: No cases of pregnancy occurred during the Skudexa clinical development. The safety profile of Skudexa during pregnancy has not been established in the clinical studies included in this section. Data reported for dexketoprofen and tramadol as single agents should be taken into account.
Dexketoprofen: Inhibition of prostaglandin synthesis may adversely affect the pregnancy and/or the embryo/foetal development. Data from epidemiological studies raise concern about an increased risk of miscarriage and of cardiac malformation and gastroschisis after use of a prostaglandin synthesis inhibitor in early pregnancy. The absolute risk for cardiovascular malformation was increased from less than 1%, up to approximately 1.5%. The risk is believed to increase with dose and duration of therapy. In animals, administration of a prostaglandin synthesis inhibitor has been shown to result in increased pre- and post-implantation loss and embryo-foetal lethality. In addition, increased incidences of various malformations including cardiovascular, have been reported in animals given a prostaglandin synthesis inhibitor during the organogenetic period. Nevertheless, animal studies with dexketoprofen haven't shown reproductive toxicity (see Pharmacology: Toxicology: Preclinical safety data under Actions).
During the third trimester of pregnancy, all prostaglandin synthesis inhibitors may expose the foetus to: cardiopulmonary toxicity (with premature closure of the ductus arteriosus and pulmonary hypertension); renal dysfunction, which may progress to renal failure with oligo-hydroamniosis.
At the end of pregnancy, the mother and the neonate may be exposed to: possible prolongation of bleeding time, an anti platelet effect which may occur even at very low doses; inhibition of uterine contractions resulting in delayed or prolonged labour.
Tramadol: Animal studies with tramadol revealed at very high doses effects on organ development, ossification and neonatal mortality. Teratogenic effects were not observed. Tramadol crosses the placenta. There is inadequate evidence available on the safety of tramadol in human pregnancy.
Tramadol - administered before or during birth - does not affect uterine contractility. In neonates it may induce changes in the respiratory rate which are usually not clinically relevant. Chronic use during pregnancy may lead to neonatal withdrawal symptoms.
Considering the previously mentioned information Skudexa is contraindicated in pregnancy (see Contraindications).
Breastfeeding: No controlled trials have been conducted to study the excretion of Skudexa in human milk. Data reported for dexketoprofen and tramadol as single agents should be taken into account.
Dexketoprofen: It is not known whether dexketoprofen is excreted in human milk.
Tramadol: Tramadol and its metabolites are found in small amounts in human breast milk.
Approximately 0.1% of the maternal dose of tramadol is excreted in breast milk. In the immediate post partum period, for maternal oral dosage up to 400 mg, this corresponds to a mean amount of tramadol ingested by breast fed infants of 3% of the maternal weight-adjusted dosage, For this reason, tramadol should not be used during lactation or alternatively, breast-feeding should be discontinued during treatment with tramadol. Discontinuation of breast-feeding is generally not necessary following a single dose of tramadol.
Considering the previously mentioned information Skudexa is contraindicated during breastfeeding (see Contraindications).
Fertility: As with other NSAIDs, the use of dexketoprofen may impair female fertility and is not recommended in women attempting to conceive. In women who have difficulties conceiving or who are undergoing investigation of infertility, withdrawal of dexketoprofen should be considered.
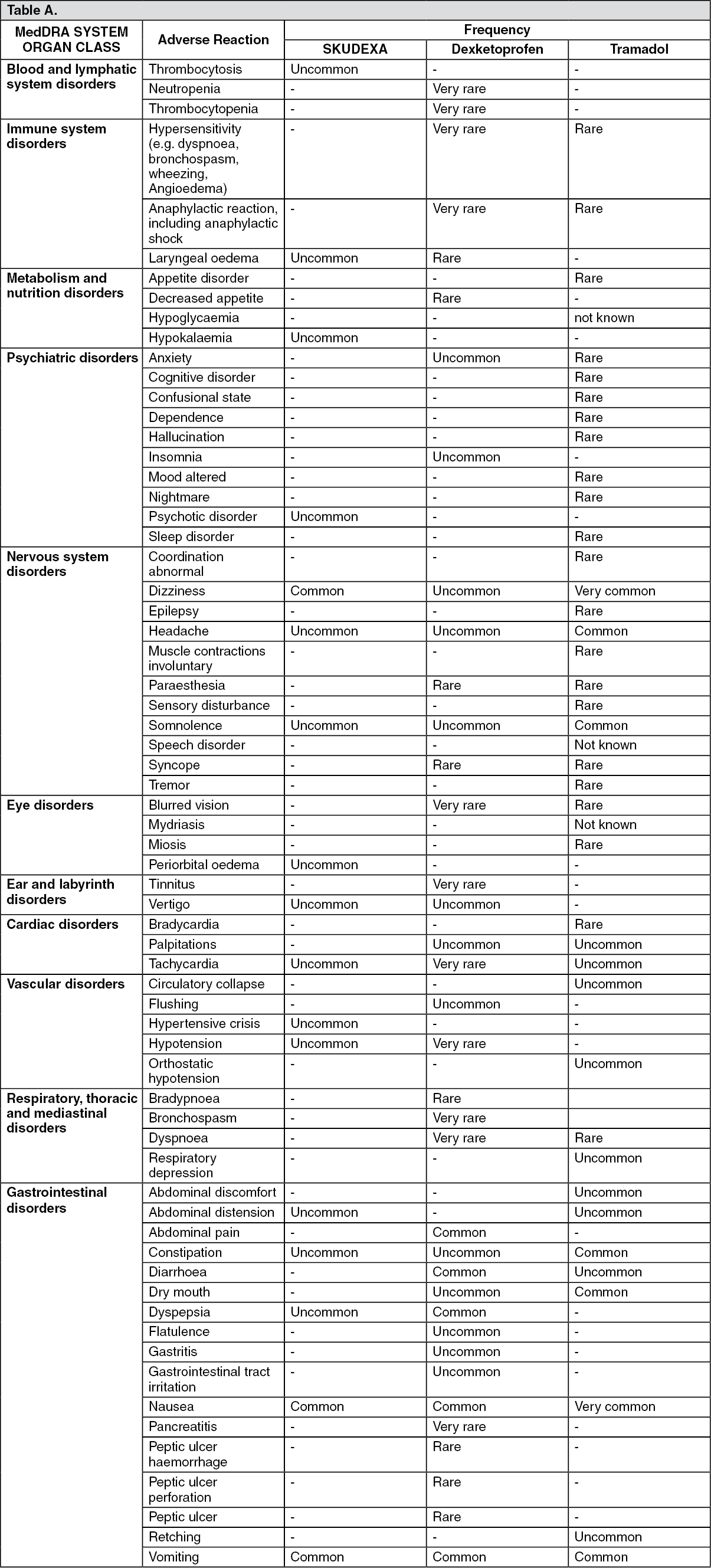
The adverse events at least possibly related reported in the clinical trials performed with Skudexa and the adverse reactions reported in dexketoprofen and tramadol oral formulations SmPCs are tabulated as follows, classified by system organ class.
The frequencies are defined as follows: Very common: ≥ 1/10; Common: ≥ 1/100 to <1/10; Uncommon: ≥1/1000 to <1/100; Rare: ≥ 1/10 000 to <1/1000; Very rare (< 1/10,000); Not known: cannot be estimated from the available data. (See table.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Dexketoprofen-tramadol:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Dexketoprofen-tramadol: In clinical studies the most commonly observed adverse reactions were vomiting, nausea and dizziness (2.9%, 2.7% and 1.1% of patients, respectively).
Dexketoprofen: Gastrointestinal: The most commonly-observed adverse events are gastrointestinal in nature. Peptic ulcers, perforation or gastrointestinal bleeding, sometimes fatal, particularly in the elderly, may occur (see Precautions). Nausea, vomiting, diarrhoea, flatulence, constipation, dyspepsia, abdominal pain, melaena, haematemesis, ulcerative stomatitis, exacerbation of colitis and Crohn's disease (see Precautions) have been reported following administration. Less frequently, gastritis has been observed. Oedema, hypertension and cardiac failure have been reported in association with NSAIDs treatment.
As with other NSAIDs the following undesirable effects may appear aseptic meningitis, which might predominantly occur in patients with systemic lupus erythematosus or mixed connective tissue disease; haematological reactions (purpura, aplastic and haemolytic anaemia, and rarely agranulocytosis and medullar hypoplasia).
Bullous reactions including Stevens Johnson Syndrome and Toxic Epidermal Necrolysis (very rare).
Clinical trial and epidemiological data suggest that use of some NSAIDs (particularly at high doses and in long term treatment) may be associated with a small increase in the risk of arterial thrombotic events (for example myocardial infarction or stroke) (see Precautions).
Tramadol: The most commonly reported adverse reactions due to tramadol are nausea and dizziness, both occurring in more than 10% of patients.
If the recommended doses are considerably exceeded and other centrally depressant substances are administered concomitantly (see Interactions) respiratory depression may occur.
Worsening of asthma has been reported, though a causal relationship has not been established.
Epileptiform convulsions occurred mainly after administration of high doses of tramadol or after concomitant treatment with drugs, which can lower the seizure threshold or themselves induce cerebral convulsions (see Precautions and Interactions).
Symptoms of withdrawal reactions, similar to those occurring during opiate withdrawal, may occur as follows; agitation, anxiety, nervousness, insomnia, hyperkinesia, tremor and gastrointestinal symptoms.
Other symptoms that have very rarely been seen with tramadol discontinuation include: panic attacks, severe anxiety, hallucinations, paraesthesias, tinnitus, and unusual CNS symptoms (i.e. confusion, delusions, depersonalisation, derealisation, paranoia).
No clinical studies have been performed to evaluate the potential impact of drug-drug interactions on safety profile of Skudexa. However, those reported for dexketoprofen and tramadol as single agents should be taken into account.
Dexketoprofen: The following interactions apply to non-steroidal anti-inflammatory drugs (NSAIDs) in general: Concomitant use not recommended: Other NSAIDs (including cyclooxygenase-2 selective inhibitors) including high doses of salicylates (≥ 3 g/day): administration of several NSAIDs together may increase the risk of gastrointestinal ulcers and bleeding, via a synergistic effect.
Anticoagulants: NSAIDs may enhance the effects of anti-coagulants, such as warfarin, due to the high plasma protein binding of dexketoprofen and the inhibition of platelet function and damage to the gastroduodenal mucosa. If the combination cannot be avoided, close clinical observation and monitoring of laboratory values should be carried out.
Heparins: increased risk of haemorrhage (due to the inhibition of platelet function and damage to the gastroduodenal mucosa). If the combination cannot be avoided, close clinical observation and monitoring of laboratory values should be carried out.
Corticosteroids: there is an increased risk of gastrointestinal ulceration or bleeding.
Lithium (described with several NSAIDs): NSAIDs increase blood lithium levels, which may reach toxic values (decreased renal excretion of lithium). This parameter therefore requires monitoring during the initiation, adjustment and withdrawal of treatment with dexketoprofen.
Methotrexate, used at high doses of 15 mg/week or more: increased haematological toxicity of methotrexate via a decrease in its renal clearance by anti-inflammatory agents in general.
Hydantoins (including phenytoin) and sulphonamides: the toxic effects of these substances may be increased.
Combinations requiring precautions: Diuretics, Angiotensin-converting-enzyme (ACE) inhibitors, antibacterial aminoglycosides and angiotensin II receptor antagonists: dexketoprofen may reduce the effect of diuretics and antihypertensive drugs. In some patients with compromised renal function (e. g. dehydrated patients or elderly patients with compromised renal function), the coadministration of agents that inhibit cyclo-oxygenase and ACE inhibitors, angiotensin II receptor antagonists or antibacterial aminoglycosides may result in further deterioration of renal function, which is usually reversible. In case of combined prescription of dexketoprofen and a diuretic, it is essential to ensure that the patient is adequately hydrated and to monitor renal function at the start of the treatment and periodically thereafter. Co-administration of dexketoprofen and potassium-sparing diuretics can lead to hyperkalaemia. Monitoring of blood potassium concentrations is required (see Precautions).
Methotrexate, used at low doses, less than 15 mg/week: increased haematological toxicity of methotrexate via a decrease in its renal clearance by anti-inflammatory agents in general. Weekly monitoring of blood count during the first weeks of the combination. Increased surveillance in the presence of even mildly impaired renal function, as well as in the elderly.
Pentoxyfilline: increased risk of bleeding. Increase clinical monitoring and check bleeding time more often.
Zidovudine: risk of increased red cell line toxicity via action on reticulocytes, with severe anaemia occurring one week after the NSAID is started. Check complete blood count and reticulocyte count one to two weeks after starting treatment with the NSAID.
Sulfonylureas: NSAIDs can increase the hypoglycaemic effect of sulfonylureas by displacement from plasma protein binding sites.
Combinations needing to be taken into account: Beta-blockers: treatment with a NSAID may decrease their antihypertensive effect via inhibition of prostaglandin synthesis.
Cyclosporin and tacrolimus: nephrotoxicity may be enhanced by NSAIDs via renal prostaglandin mediated effects. During combination therapy, renal function has to be measured.
Thrombolytics: increased risk of bleeding.
Anti-platelet agents and selective serotonin reuptake inhibitors (SSRIs): increased risk of gastrointestinal bleeding (see Precautions).
Probenecid: plasma concentrations of dexketoprofen may be increased; this interaction can be due to an inhibitory mechanism at the site of renal tubular secretion and of glucuronoconjugation and requires adjustment of the dose of dexketoprofen.
Cardiac glycosides: NSAIDS may increase plasma glycoside concentration.
Mifepristone: because of a theoretical risk that prostaglandin synthetase inhibitors may alter the efficacy of mifepristone, NSAIDS should not be used for 8-12 days after mifepristone administration.
Limited evidence suggests that co-administration of NSAIDs on the day of prostaglandin administration does not adversely influence the effects of mifepristone or the prostaglandin on cervical ripening or uterine contractility and does not reduce the clinical efficacy of medical termination of pregnancy.
Quinolone antibiotics: animal data indicate that high doses of quinolones in combination with NSAIDS can increase the risk of developing convulsions.
Tenofovir: concomitant use with NSAID can increase plasma urea nitrogen and creatinine, renal function should be monitored in order to control a potential synergic influence on renal function.
Deferasirox: concomitant use with NSAIDs can increase the risk of gastrointestinal toxicity. Close clinical monitoring is required when deferasirox is combined with these substances.
Pemetrexed: concomitant use with NSAIDs may decrease pemetrexed elimination, therefore caution should be made when administering higher doses of NSAIDs. In patients with mild to moderate renal insufficiency (creatinine clearance from 45 to 79 ml/min), the concomitant administration of pemetrexed with NSAIDs doses should be avoided for 2 days before and 2 days following pemetrexed administration.
Tramadol: Concomitant use not recommended: Tramadol should not be combined with Monoamine Oxidase (MAO) inhibitors (see Contraindications). In patients treated with MAO inhibitors in the 14 days prior to the use of the opioid pethidine, life-threatening interactions on the central nervous system, respiratory and cardiovascular function have been observed. The same interactions with MAO inhibitors cannot be ruled out during treatment with tramadol.
Caution should be exercised during concomitant treatment with tramadol and coumarin derivatives (e.g. warfarin) due to reports of elevated International Normalized Ratio (INR) with major bleeding and ecchymoses in some patients.
The combination of mixed agonists/antagonists opioid receptors (e.g. buprenorphine, nalbuphine, pentazocine) and tramadol is not advisable because the analgesic effect of a pure agonist may be theoretically reduced in such circumstances.
Combinations requiring precautions: Tramadol can induce convulsions and increase the potential for selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants, antipsychotics and other seizure threshold-lowering medicinal product (such as bupropion, mirtazapine, tetrahydrocannabinol) to cause convulsions.
Concomitant therapeutic use of tramadol and serotonergic drugs, such as selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), MAO inhibitors (see Contraindications), tricyclic antidepressants and mirtazapine may cause serotonin toxicity. Serotonin syndrome is likely when one of the following is observed: spontaneous clonus, inducible or ocular clonus with agitation or diaphoresis, tremor and hyperreflexia, hypertonia and body temperature > 38°C and inducible ocular clonus. Withdrawal of the serotonergic drugs usually brings about a rapid improvement. Treatment depends on the type and severity of the symptoms.
The concomitant use of opioids with sedative medicines such as benzodiazepines or related drugs increases the risk of sedation, respiratory depression, coma and death because of additive CNS depressant effect. The dose and duration of concomitant use should be limited (see Precautions).
Combinations needing to be taken into account: Concomitant administration of tramadol with other centrally depressant medicinal products or alcohol may potentiate the central nervous system effects (see Adverse Reactions).
The results of pharmacokinetic studies have so far shown that on the concomitant or previous administration of cimetidine (enzyme inhibitor) clinically relevant interactions are unlikely to occur.
Simultaneous or previous administration of carbamazepine (enzyme inducer) may reduce the analgesic effect and shorten the duration of action.
In a limited number of studies the pre- or postoperative administration of the antiemetic 5-HT3 antagonist ondansetron increased the requirement of tramadol in patients with postoperative pain.
Other active substances known to inhibit CYP3A4, such as ketoconazole and erythromycin, might inhibit the metabolism of tramadol (N-demethylation) probably also the metabolism of the active O-demethylated metabolite. The clinical importance of such an interaction has not been studied.
Incompatibilities: Not applicable.
Special precautions for disposal: No special requirements.
Any unused product or waste material should be disposed of in accordance with local requirements.
Store below 30°C in the original package in order to protect from light.
Shelf life: 48 months.
N02AJ14 - tramadol and dexketoprofen ; Belongs to the class of opioids in combination with other non-opioid analgesics. Used to relieve pain.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image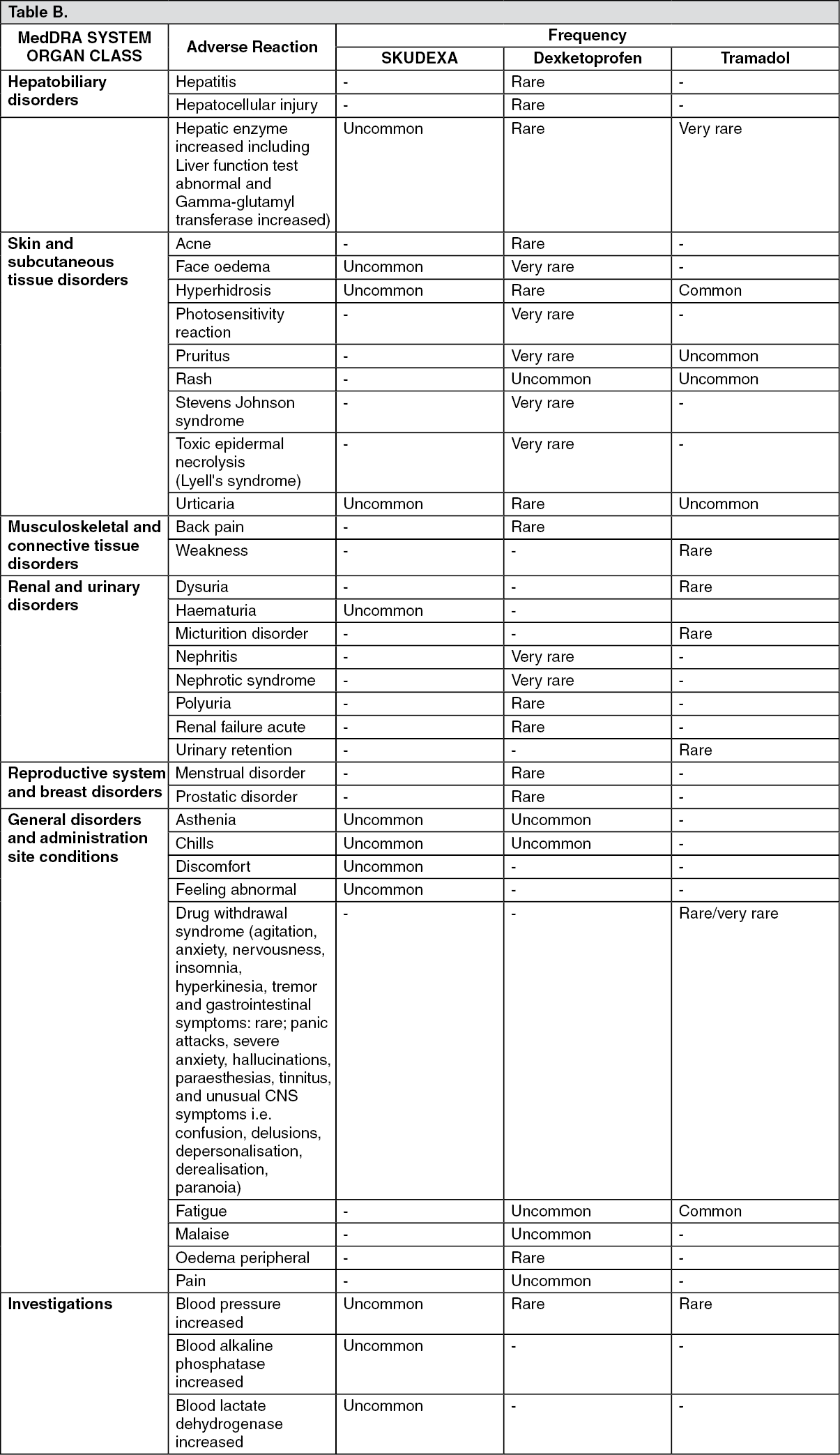 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
