Pharmacology: Mechanism of Action: Tedizolid phosphate is the prodrug of tedizolid, an antibacterial agent [see Pharmacokinetics and Microbiology as follows].
Pharmacodynamics: The AUC/minimum inhibitory concentration (MIC) was shown to best correlate with tedizolid activity in animal infection models.
In the mouse thigh infection model of
S. aureus, antistaphylococcal killing activity was impacted by the presence of granulocytes. In granulocytopenic mice (neutrophil count <100 cells/mL), bacterial stasis was achieved at a human-equivalent dose of approximately 2000 mg/day; whereas, in non-granulocytopenic animals, stasis was achieved at a human-equivalent dose of approximately 100 mg/day. The safety and efficacy of SIVEXTRO for the treatment of neutropenic patients (neutrophil counts <1000 cells/mm
3) have not been evaluated.
Cardiac Electrophysiology: In a randomized, positive- and placebo-controlled crossover thorough QTc study, 48 enrolled subjects were administered a single oral dose of SIVEXTRO at a therapeutic dose of 200 mg, SIVEXTRO at a supratherapeutic dose of 1200 mg, placebo, and a positive control; no significant effects of SIVEXTRO on heart rate, electrocardiogram morphology, PR, QRS, or QT interval were detected. Therefore, SIVEXTRO does not affect cardiac repolarization.
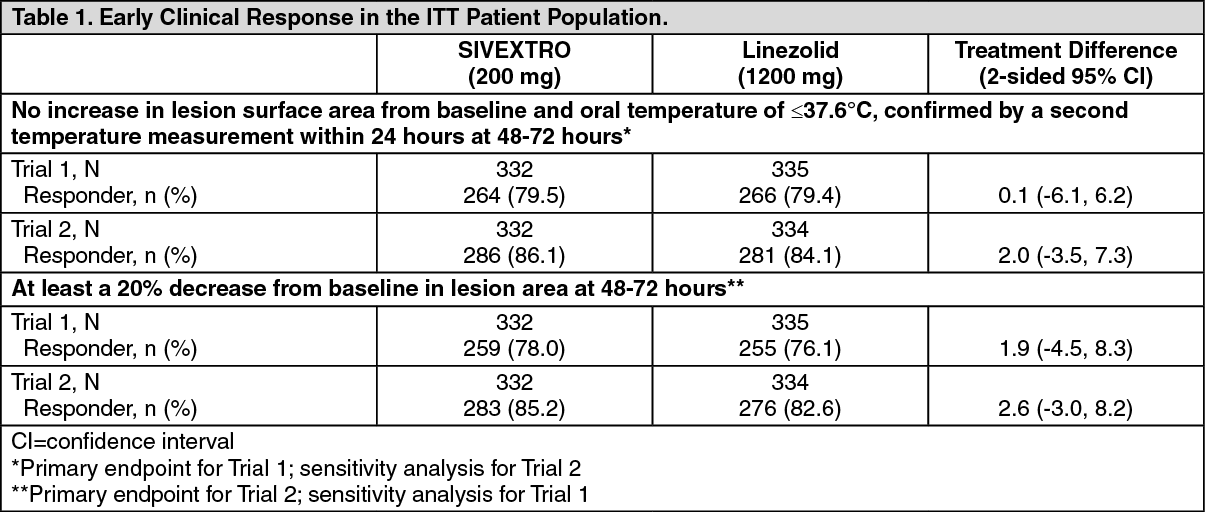
Clinical Studies: Acute Bacterial Skin and Skin Structure Infections: A total of 1335 adults with acute bacterial skin and skin structure infections (ABSSSI) were randomized in two multicenter, multinational, double-blind, non-inferiority trials. Both trials compared SIVEXTRO 200 mg once daily for 6 days versus linezolid 600 mg every 12 hours for 10 days. In Trial 1, patients were treated with oral therapy, while in Trial 2, patients could receive oral therapy after a minimum of one day of intravenous therapy. Patients with cellulitis/erysipelas, major cutaneous abscess, or wound infection were enrolled in the trials. Patients with wound infections could have received aztreonam and/or metronidazole as adjunctive therapy for gram-negative bacterial coverage, if needed. The intent-to-treat (ITT) patient population included all randomized patients.
In Trial 1, 332 patients with ABSSSI were randomized to SIVEXTRO and 335 patients were randomized to linezolid. The majority (91%) of patients treated with SIVEXTRO in Trial 1 were less than 65 years old with a median age of 43 years (range: 18 to 86 years). Patients treated with SIVEXTRO were predominantly male (61%) and White (84%); 13% had BMI ≥35 kg/m
2, 8% had diabetes mellitus, 35% were current or recent intravenous drug users, and 2% had moderate to severe renal impairment. The overall median surface area of infection was 188 cm
2. The types of ABSSSI included were cellulitis/erysipelas (41%), wound infection (29%), and major cutaneous abscess (30%). In addition to local signs and symptoms of infection, patients were also required to have at least one regional or systemic sign of infection at baseline, defined as lymphadenopathy (87% of patients), temperature 38°C or higher (16% of patients), white blood cell count greater than 10,000 cells/mm
3 or less than 4000 cells/mm
3 (42%), or 10% or more band forms on white blood cell differential (4%).
The primary endpoint in Trial 1 was early clinical response defined as no increase from baseline lesion area at 48-72 hours after the first dose and oral temperature of ≤37.6°C, confirmed by a second temperature measurement within 24 hours in the ITT population.
In Trial 2, 332 patients with ABSSSI were randomized to SIVEXTRO and 334 patients were randomized to linezolid. The majority (87%) of patients treated with SIVEXTRO in Trial 2 were less than 65 years old with a median age of 46 years (range: 17 to 86 years). Patients treated with SIVEXTRO were predominantly male (68%) and White (86%); 16% had BMI ≥35 kg/m
2, 10% had diabetes mellitus, 20% were current or recent intravenous drug users, and 4% had moderate to severe renal impairment. The overall median surface area of infection was 231 cm
2. The types of ABSSSI included were cellulitis/erysipelas (50%), wound infection (30%), and major cutaneous abscess (20%). In addition to local signs and symptoms of infection, patients were also required to have at least one regional or systemic sign of infection at baseline, defined as lymphadenopathy (71% of patients), temperature 38°C or higher (31% of patients), white blood cell count greater than 10,000 cells/mm
3 or less than 4000 cells/mm
3 (53%), or 10% or more band forms on white blood cell differential (16%).
The primary endpoint in Trial 2 was early clinical response defined as at least a 20% decrease from baseline lesion area at 48-72 hours after the first dose in the ITT population (see Table 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
An investigator assessment of clinical response was made at the post-therapy evaluation (PTE) (7-14 days after the end of therapy) in the ITT and CE (Clinically Evaluable) populations. Clinical success was defined as resolution or near resolution of most disease-specific signs and symptoms, absence or near resolution of systemic signs of infection if present at baseline (lymphadenopathy, fever, >10% immature neutrophils, abnormal WBC count), and no new signs, symptoms, or complications attributable to the ABSSSI requiring further treatment of the primary lesion (see Table 2.)
Click on icon to see table/diagram/image
Clinical success by baseline pathogens from the primary infection site or blood cultures for the microbiological intent-to-treat (MITT) patient population for two integrated Phase 3 ABSSSI studies are presented in Table 3 and Table 4. (See Table 3.)
Click on icon to see table/diagram/image
Baseline bacteremia in the tedizolid arm with relevant pathogens included two subjects with MRSA, four subjects with MSSA, two subjects with
S. pyogenes, one subject with
S. agalactiae, and one subject with
S. constellatus. All of these subjects were Responders at the 48-72 hour evaluation. At the Post-therapy Evaluation (PTE), 8 of 10 subjects were considered clinical successes. (See Table 4.)
Click on icon to see table/diagram/image
Baseline bacteremia in the tedizolid arm with relevant pathogens included two subjects with MRSA, four subjects with MSSA, two subjects with
S. pyogenes, one subject with
S.agalactiae, and one subject with
S. constellatus. All of these subjects were Responders at the 48-72 hour evaluation. At the Post-therapy Evaluation (PTE) 8 of 10 subjects were considered clinical successes.
Pharmacokinetics: Tedizolid phosphate is a prodrug that is converted by phosphatases to tedizolid, the microbiologically active moiety, following oral and intravenous administration. Only the pharmacokinetic profile of tedizolid is discussed further due to negligible systemic exposure of tedizolid phosphate following oral and intravenous administration. Following multiple once-daily oral or intravenous administration, steady-state concentrations are achieved within approximately three days with tedizolid accumulation of approximately 30% (tedizolid half-life of approximately 12 hours). Pharmacokinetic (PK) parameters of tedizolid following oral and intravenous administration of 200 mg once daily tedizolid phosphate are shown in Table 5. (See Table 5.)
Click on icon to see table/diagram/image
Absorption: Peak plasma tedizolid concentrations are achieved within approximately 3 hours following oral administration under fasting conditions or at the end of the 1 hour intravenous infusion of tedizolid phosphate. The absolute bioavailability is approximately 91% and no dosage adjustment is necessary between intravenous and oral administration.
Tedizolid phosphate (oral) may be administered with or without food as total systemic exposure (AUC
0-∞) is unchanged between fasted and fed (high-fat, high-calorie) conditions.
Distribution: Protein binding of tedizolid to human plasma proteins is approximately 70 to 90%. The mean steady state volume of distribution of tedizolid in healthy adults following a single intravenous dose of tedizolid phosphate 200 mg ranged from 67 to 80 L (approximately twice total body water). Tedizolid penetrates into the interstitial space fluid of adipose and skeletal muscle tissue with exposure similar to free drug exposure in plasma.
Metabolism: Other than tedizolid, which accounts for approximately 95% of the total radiocarbon AUC in plasma, there are no other significant circulating metabolites in humans.
There was no degradation of tedizolid in human liver microsomes indicating tedizolid is unlikely to be a substrate for hepatic CYP450 enzymes.
In vitro studies showed that conjugation of tedizolid is mediated via multiple sulfotransferase (SULT) isoforms (SULT1 A1, SULT1 A2, SULT2 A1).
Excretion: Following single oral administration of
14C-labeled tedizolid phosphate under fasted conditions, the majority of elimination occurred via the liver, with 82% of the radioactive dose recovered in feces and 18% in urine, primarily as a non-circulating and microbiologically inactive sulfate conjugate. Most of the elimination of tedizolid (>85%) occurs within 96 hours. Less than 3% of the tedizolid phosphate-administered dose is excreted in feces and urine as unchanged tedizolid.
Specific Populations: Based on the population pharmacokinetic analysis, there are no clinically relevant demographic or clinical patient factors (including age, gender, race, ethnicity, weight, body mass index, and measures of renal or liver function) that impact the pharmacokinetics of tedizolid.
Hepatic Impairment: Following administration of a single 200 mg oral dose of SIVEXTRO, no clinically meaningful changes in mean tedizolid C
max and AUC
0-∞ were observed in patients with moderate (n=8) or severe (n=8) hepatic impairment (Child-Pugh Class B and C) compared to 8 matched healthy control subjects. No dose adjustment is necessary for patients with hepatic impairment.
Renal Impairment: Following administration of a single 200 mg intravenous dose of SIVEXTRO to 8 subjects with severe renal impairment defined as eGFR <30 mL/min/1.73 m
2, the C
max was essentially unchanged and AUC
0-∞ was decreased by less than 10% compared to 8 matched healthy control subjects. Hemodialysis does not result in meaningful removal of tedizolid from systemic circulation, as assessed in subjects with end-stage renal disease (eGFR <15 mL/min/1.73 m
2). No dosage adjustment is necessary in patients with renal impairment or patients on hemodialysis.
Geriatric Patients: The pharmacokinetics of tedizolid were evaluated in a Phase 1 study conducted in elderly healthy volunteers (age 65 years and older, with at least 5 subjects at least 75 years old; n=14) compared to younger control subjects (25 to 45 years old; n=14) following administration of a single oral dose of SIVEXTRO 200 mg. There were no clinically meaningful differences in tedizolid C
max and AUC
0-∞ between elderly subjects and younger control subjects. No dosage adjustment of SIVEXTRO is necessary in elderly patients.
Gender: The impact of gender on the pharmacokinetics of SIVEXTRO was evaluated in clinical trials of healthy males and females and in a population pharmacokinetics analysis. The pharmacokinetics of tedizolid were similar in males and females. No dosage adjustment of SIVEXTRO is necessary based on gender.
Drug Interaction Studies: Drug Metabolizing Enzymes: Transformation via Phase 1 hepatic oxidative metabolism is not a significant pathway for elimination of SIVEXTRO.
Neither SIVEXTRO nor tedizolid detectably inhibited or induced the metabolism of selected CYP enzyme substrates, suggesting that drug-drug interactions based on oxidative metabolism are unlikely.
Membrane Transporters: The potential for tedizolid or tedizolid phosphate to inhibit transport of probe substrates of important drug uptake (OAT1, OAT3, OATP1B1, OATP1B3, OCT1, and OCT2) and efflux transporters (P-gp and BCRP) was tested
in vitro. No clinically relevant interactions are expected to occur with these transporters except BCRP.
Coadministration of multiple oral doses of SIVEXTRO (200 mg once daily) increased the C
max and AUC of rosuvastatin (10 mg single oral dose), a known BCRP substrate, by approximately 55% and 70%, respectively, in healthy subjects [see Interactions].
Monoamine Oxidase Inhibition: Tedizolid is a reversible inhibitor of monoamine oxidase (MAO)
in vitro. The interaction with MAO inhibitors could not be evaluated in Phase 2 and 3 trials, as subjects taking such medications were excluded from the trials.
Adrenergic Agents: Two placebo-controlled crossover studies were conducted to assess the potential of 200 mg oral SIVEXTRO at steady state to enhance pressor responses to pseudoephedrine and tyramine in healthy individuals. No meaningful changes in blood pressure or heart rate were seen with pseudoephedrine. The median tyramine dose required to cause an increase in systolic blood pressure of ≥30 mmHg from pre-dose baseline was 325 mg with SIVEXTRO compared to 425 mg with placebo. Palpitations were reported in 21/29 (72.4%) subjects exposed to SIVEXTRO compared to 13/28 (46.4%) exposed to placebo in the tyramine challenge study.
Serotonergic Agents: Serotonergic effects at doses of tedizolid phosphate up to 30-fold above the human equivalent dose did not differ from vehicle control in a mouse model that predicts serotonergic activity. In Phase 3 trials, subjects taking serotonergic agents including antidepressants such as selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants, and serotonin 5-hydroxytryptamine (5-HT1) receptor agonists (triptans), meperidine, or buspirone were excluded.
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Long-term carcinogenicity studies have not been conducted with tedizolid phosphate.
Tedizolid phosphate was negative for genotoxicity in all
in vitro assays (bacterial reverse mutation (Ames), Chinese hamster lung (CHL) cell chromosomal aberration) and in all
in vivo tests (mouse bone marrow micronucleus, rat liver unscheduled DNA synthesis). Tedizolid, generated from tedizolid phosphate after metabolic activation (
in vitro and
in vivo), was also tested for genotoxicity. Tedizolid was positive in an
in vitro CHL cell chromosomal aberration assay, but negative for genotoxicity in other
in vitro assays (Ames, mouse lymphoma mutagenicity) and
in vivo in a mouse bone marrow micronucleus assay.
In a fertility study, oral tedizolid phosphate had no adverse effects on the fertility or reproductive performance, including spermatogenesis, of male rats at the maximum tested dose (50 mg/kg/day) with a plasma tedizolid AUC approximately 5-fold greater than the plasma AUC value in humans at the oral therapeutic dose. Tedizolid phosphate also had no adverse effects on the fertility or reproductive performance of adult female rats at doses up to the maximum tested (15 mg/kg/day). Plasma tedizolid exposure (AUC) at this NOAEL in female rats was approximately 4-fold higher than that in humans at the oral therapeutic dose.
Animal Toxicology and/or Pharmacology: Repeated-oral and intravenous dosing of tedizolid phosphate in rats in 1-month and 3-month toxicology studies produced dose- and time-dependent bone marrow hypocellularity (myeloid, erythroid, and megakaryocyte), with associated reduction in circulating RBCs, WBCs, and platelets. These effects showed evidence of reversibility and occurred at plasma tedizolid exposure levels (AUC) ≥6-fold greater than the plasma exposure associated with the human therapeutic dose. In a 1-month immunotoxicology study in rats, repeated oral dosing of tedizolid phosphate was shown to significantly reduce splenic B cells and T cells and reduce plasma IgG titers. These effects occurred at plasma tedizolid exposure levels (AUC) ≥3-fold greater than the expected human plasma exposure associated with the therapeutic dose.
Microbiology: Tedizolid belongs to the oxazolidinone class of antibacterial drugs.
Mechanism of Action: The antibacterial activity of tedizolid is mediated by binding to the 50S subunit of the bacterial ribosome resulting in inhibition of protein synthesis. Tedizolid inhibits bacterial protein synthesis through a mechanism of action different from that of other non-oxazolidinone class antibacterial drugs; therefore, cross-resistance between tedizolid and other classes of antibacterial drugs is unlikely. The results of
in vitro time-kill studies show that tedizolid is bacteriostatic against enterococci, staphylococci, and streptococci.
Mechanism of Resistance: Organisms resistant to oxazolidinones via mutations in chromosomal genes encoding 23S rRNA or ribosomal proteins (L3 and L4) are generally cross-resistant to tedizolid. In the limited number of
Staphylococcus aureus strains tested, the presence of the chloramphenicol-florfenicol resistance (cfr) gene did not result in resistance to tedizolid in the absence of chromosomal mutations.
Frequency of Resistance: Spontaneous mutations conferring reduced susceptibility to tedizolid occur
in vitro at a frequency rate of approximately 10
-10.
In experiments to evaluate the resistance development during serial passage, the potential for the development of resistance to tedizolid appears to be equivalent to or less than that of linezolid. In a direct comparison of
S. aureus ATCC 29213 (MSSA) and
S. aureus ATCC 33591 (MRSA), tedizolid displayed an increase of MIC of zero- and 8-fold whereas linezolid displayed an increase of MIC of 64- and 32-fold, respectively.
Results of a large global survey of 36,573 clinical isolates of
S. aureus tested over a 5-year period from 2015-2019 showed that only a single clinical isolate of MRSA was non-susceptible to oxazolidinones. This
S. aureus isolate showed the G2576T alteration in all 6 23S rRNA alleles, which affects oxazolidinone binding and explains the elevated MIC values observed for tedizolid (8 mg/L) and linezolid (32 mg/L). No new mechanisms of resistance were observed in this global survey. All other
S. aureus isolates were inhibited by tedizolid at MIC values of ≤ 0.5 mg/L.
Interaction with Other Antimicrobial Drugs: In vitro drug combination studies with tedizolid and aztreonam, ceftriaxone, ceftazidime, imipenem, rifampin, trimethoprim/sulfamethoxazole, minocycline, clindamycin, ciprofloxacin, daptomycin, vancomycin, gentamicin, amphotericin B, ketoconazole, and terbinafine demonstrate neither synergy nor antagonism.
Spectrum of Activity: Tedizolid has been shown to be active against most isolates of the following bacteria, both
in vitro and in clinical infections, as described in Indications.
Aerobic and Facultative Gram-positive Bacteria: Staphylococcus aureus (including methicillin-resistant [MRSA] and methicillin-susceptible [MSSA] isolates),
Streptococcus pyogenes,
Streptococcus agalactiae,
Streptococcus anginosus Group (including
S. anginosus,
S. intermedius, and
S. constellatus),
Enterococcus faecalis.
The following
in vitro data are available, but their clinical significance has not been established. At least 90% of the following microorganisms exhibit an
in vitro minimum inhibitory concentration (MIC) less than or equal to 0.5 mcg/mL for tedizolid. However, the safety and effectiveness of SIVEXTRO in treating clinical infections due to these microorganisms have not been established in adequate and well-controlled clinical trials.
Aerobic and Facultative Anaerobic Gram-positive Bacteria: Staphylococcus epidermidis (including methicillin-susceptible and methicillin-resistant isolates),
Staphylococcus haemolyticus,
Staphylococcus lugdunensis,
Enterococcus faecium.
Susceptibility Test Methods: When available, the clinical microbiology laboratory should provide cumulative results of the
in vitro susceptibility test results for antimicrobial drugs used in local hospitals and practice areas to the physician as periodic reports that describe the susceptibility profile of nosocomial and community-acquired pathogens. These reports should aid the physician in selecting an effective antibacterial drug for treatment.
Dilution Techniques: Quantitative methods are used to determine antimicrobial minimum inhibitory concentrations (MICs). These MIC values provide estimates of the susceptibility of bacteria to antimicrobial compounds. The MIC values should be determined using a standardized procedure based on dilution methods (broth, agar, or microdilution) or equivalent using standardized inoculum and concentrations of tedizolid. The MIC values should be interpreted according to the criteria provided in Table 6. (See Table 6.)
Click on icon to see table/diagram/image
Diffusion techniques: Quantitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. The standardized procedure requires the use of standardized inoculum concentrations. This procedure uses paper disks impregnated with 20 mcg tedizolid to test the susceptibility of microorganisms to tedizolid. Reports from the laboratory providing results of the standard single-disk susceptibility test with a 20 mcg tedizolid disk should be interpreted according to the criteria in Table 6.
A report of "Susceptible" indicates that the antimicrobial drug is likely to inhibit growth of the pathogen if the antimicrobial drug reaches the concentration usually achievable at the site of infection. A report of "Intermediate" indicates that the result should be considered equivocal, and if the microorganism is not fully susceptible to alternative drugs, the test should be repeated. This category implies possible clinical efficacy in body sites where the drug is physiologically concentrated. This category also provides a buffer zone that prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of "Resistant" indicates that the antimicrobial drug is not likely to inhibit growth of the pathogen if the antimicrobial drug reaches the concentrations usually achievable at the infection site; other therapy should be selected.
Quality Control: Standardized susceptibility test procedures require the use of laboratory control microorganisms to monitor and ensure the accuracy and precision of supplies and reagents used in the assay, and the techniques of the individuals performing the test. Standardized tedizolid powder should provide the following range of MIC values noted in Table 7. For the diffusion technique using the 20 mcg tedizolid disk, results within the ranges specified in Table 7 should be observed. (See Table 7.)
Click on icon to see table/diagram/image



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image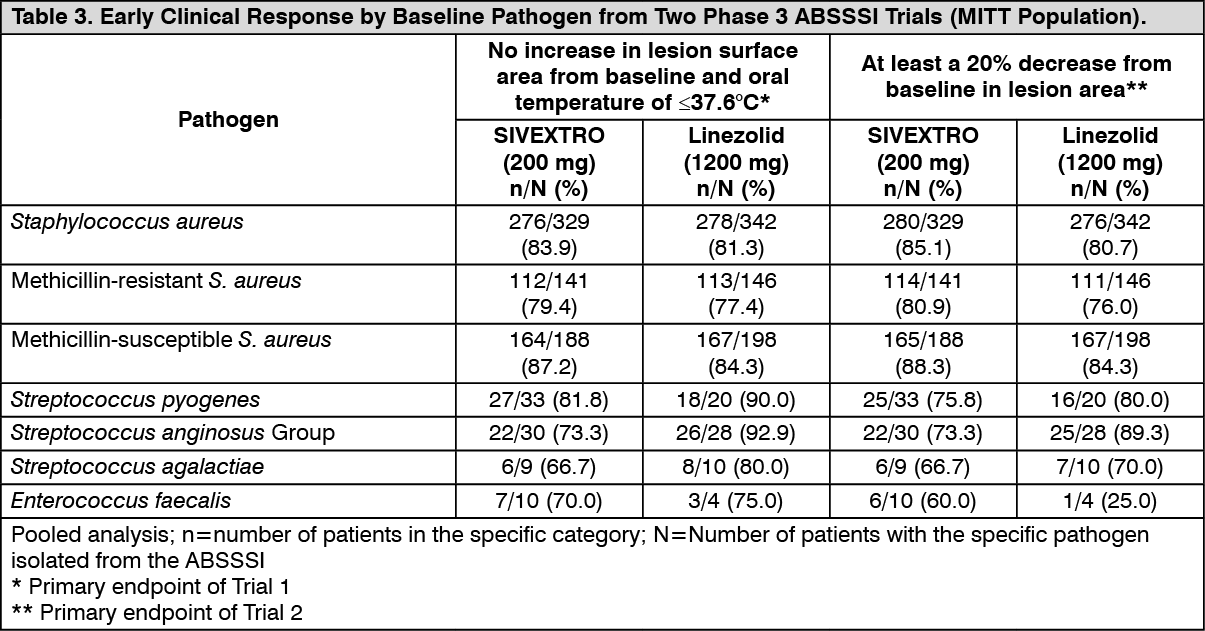 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image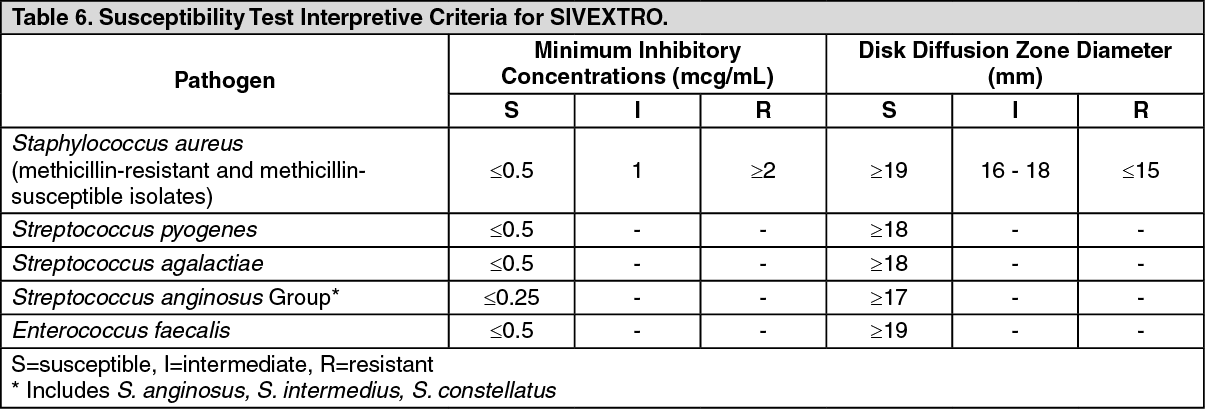 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image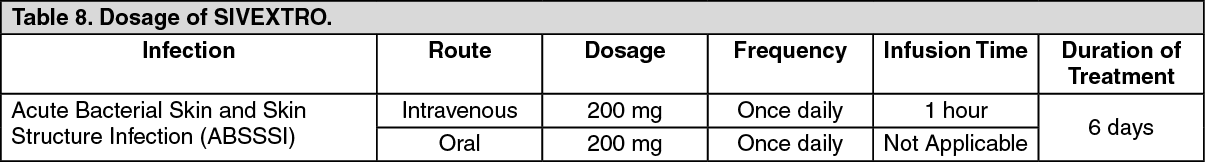 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image 200 mga413e647-f7fe-42c9-8d3e-ae7e00ddb09a.GIF)

 Sign Out
Sign Out
