Pharmacotherapeutic group: Immunosuppressants, tumour necrosis factor alpha (TNF-α) inhibitors.
ATC code: L04AB06.
Pharmacology: Pharmacodynamics: Mechanism of action: Golimumab is a human monoclonal antibody that forms high affinity, stable complexes with both the soluble and transmembrane bioactive forms of human TNF, which prevents the binding of TNF-α to its receptors.
Pharmacodynamic effects: The binding of human TNF by golimumab was shown to neutralise TNF-α-induced cell-surface expression of the adhesion molecules E-selectin, vascular cell adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1 by human endothelial cells.
In-vitro, TNF-induced secretion of interleukin (IL)-6, IL-8 and granulocyte-macrophage colony stimulating factor (GM-CSF) by human endothelial cells was also inhibited by golimumab.
Improvement in C-reactive protein (CRP) levels were observed relative to placebo groups and treatment with SIMPONI resulted in significant reductions from baseline in serum levels of IL-6, ICAM-1, matrix metalloproteinase-3 (MMP-3) and vascular endothelial growth factor (VEGF) compared to control treatment. In addition, levels of TNF-α were reduced in RA and AS patients and levels of IL-8 were reduced in PsA patients. These changes were observed at the first assessment (week 4) after the initial SIMPONI administration and were generally maintained through week 24.
Clinical efficacy: Rheumatoid arthritis: The efficacy of SIMPONI was demonstrated in three multi-centre, randomised, double-blind, placebo-controlled studies in over 1500 patients ≥ 18 years of age with moderately to severely active RA diagnosed according to American College of Rheumatology (ACR) criteria for at least 3 months prior to screening. Patients had at least 4 swollen and 4 tender joints. SIMPONI or placebo were subcutaneously administered every 4 weeks.
GO-FORWARD evaluated 444 patients who had active RA despite a stable dose of at least 15 mg/week of MTX and who had not been previously treated with an anti-TNF agent. Patients were randomised to receive placebo + MTX, SIMPONI 50 mg + MTX, SIMPONI 100 mg + MTX or SIMPONI 100 mg + placebo. Patients receiving placebo + MTX were switched to SIMPONI 50 mg + MTX after week 24. At week 52, patients entered an open label long-term extension.
GO-AFTER evaluated 445 patients who were previously treated with one or more of the anti-TNF agents adalimumab, etanercept, or infliximab. Patients were randomised to receive placebo, SIMPONI 50 mg, or SIMPONI 100 mg. Patients were allowed to continue concomitant DMARD therapy with MTX, sulfasalazine (SSZ), and/or hydroxychloroquine (HCQ) during the study. The stated reasons for discontinuation of prior anti-TNF therapies were lack of efficacy (58%), intolerance (13%), and/or reasons other than safety or efficacy (29%, mostly for financial reasons).
GO-BEFORE evaluated 637 patients with active RA who were MTX-naïve and had not previously been treated with an anti-TNF agent. Patients were randomised to receive placebo + MTX, SIMPONI 50 mg + MTX, SIMPONI 100 mg + MTX or SIMPONI 100 mg + placebo. At week 52, patients entered an open label long-term extension in which patients receiving placebo + MTX who had at least 1 tender or swollen joint were switched to SIMPONI 50 mg + MTX.
In GO-FORWARD, the (co-)primary endpoints were the percentage of patients achieving an ACR 20 response at Week 14 and the improvement from baseline in Health Assessment Questionnaire (HAQ) at Week 24. In GO-AFTER, the primary endpoint was the percentage of patients achieving an ACR 20 response at week 14. In GO-BEFORE, the co-primary endpoints were the percentage of patients achieving ACR 50 response at Week 24 and the change from baseline in the van der Heijde-modified Sharp (vdH-S) score at week 52. In addition to the primary endpoint(s), additional assessments of the impact of SIMPONI treatment on the signs and symptoms of arthritis, radiographic response, physical function and health-related quality of life were performed.
In general, no clinically meaningful differences in measures of efficacy were observed between the SIMPONI 50 mg and 100 mg dosing regimens with concomitant MTX, through week 104 in GO-FORWARD and GO-BEFORE and through week 24 in GO-AFTER. In each of the RA studies by study design, patients in the long-term extension may have switched between the 50 mg and 100 mg SIMPONI doses at the discretion of the study physician.
IV Study RA-1 (GO-FURTHER) evaluated 592 patients with active RA despite concurrent MTX therapy. Patients were randomized to receive either SIMPONI 2 mg/kg IV (N=395) or IV placebo (saline) (N=197) at Week 0, Week 4, and every 8 weeks thereafter in addition to their weekly maintenance MTX dose. All patients receiving IV placebo + MTX received SIMPONI 2 mg/kg IV + MTX after Week 24, but the trial remained double-blind through 52 weeks of treatment. The primary endpoint was the percentage of patients achieving ACR 20 response at Week 14. The major secondary endpoints included DAS28 response (using CRP) and change from baseline in HAQ-DI at Week 14 as well as ACR 50 response and change from baseline in vdH-s score at Week 24. Other pre-specified endpoints included improvement in ACR components, ACR response over time, improvement in physical function and health-related quality of life, as well as health economics measures.
Signs and symptoms: Key ACR results for the SIMPONI 50 mg dose at weeks 14, 24 and 52 for GO-FORWARD, GO-AFTER and GO-BEFORE are shown in Table 1 and are described as follows. Responses were observed at the first assessment (Week 4) after the initial SIMPONI administration.
In GO-FORWARD, among 89 subjects randomised to SIMPONI 50 mg + MTX, 48 were still on this treatment at week 104. Among those, 40, 33 and 24 patients had ACR 20/50/70 response, respectively at week 104. Among patients remaining in the study and treated with SIMPONI, similar rates of ACR 20/50/70 response was observed from week 104 through week 256.
In GO-AFTER, the percentage of patients achieving an ACR 20 response was greater for patients receiving SIMPONI than for patients receiving placebo regardless of the reason reported for discontinuation of one or more prior anti-TNF therapies. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In GO-BEFORE the primary analysis in patients with moderate to severe rheumatoid arthritis (combined SIMPONI 50 and 100 mg + MTX groups vs MTX alone for ACR50) was not statistically significant at week 24 (p = 0.053). At week 52 in the overall population, the percentage of patients in the SIMPONI 50 mg + MTX group who achieved an ACR response was generally higher but not significantly different when compared with MTX alone (see Table 3). Additional analyses were performed in subsets representative of the indicated population of patients with severe, active and progressive RA. A generally greater effect of SIMPONI 50 mg + MTX versus MTX alone was demonstrated in the indicated population compared with the overall population.
In GO-FORWARD and GO-AFTER, clinically meaningful and statistically significant responses in Disease Activity Scale (DAS)28 were observed at each prespecified time point, at week 14 and at week 24 (p ≤ 0.001). Among patients who remained on the SIMPONI treatment to which they were randomised at study start, DAS28 responses were maintained through week 104. Among patients remaining in the study and treated with SIMPONI, DAS28 responses were similar from week 104 through week 256.
In GO-BEFORE, major clinical response, defined as the maintenance of an ACR 70 response over a continuous 6-month period, was measured. At week 52, 15% of patients in the SIMPONI 50 mg + MTX group achieved a major clinical response compared with 7% of patients in the placebo + MTX group (p = 0.018). Among 159 subjects randomised to SIMPONI 50 mg + MTX, 96 were still on this treatment at week 104. Among those, 85, 66 and 53 patients had ACR 20/50/70 response, respectively, at week 104. Among patients remaining in the study and treated with SIMPONI, similar rates of ACR 20/50/70 response were observed from week 104 through week 256.
Treatment with SIMPONI IV in patients with active RA despite MTX resulted in improvement in signs and symptoms as demonstrated by the percentage of patients achieving an ACR 20 response at Week 14. A significantly greater percentage of patients achieved an ACR 20 response in the SIMPONI IV + MTX group than in the placebo IV + MTX group (p<0.001). In the SIMPONI IV + MTX group, 58.5% of patients achieved an ACR 20 response compared with 24.9% in the placebo IV + MTX group at Week 14. The percentage of patients achieving an ACR 20 response at Week 24 was also significantly greater for patients receiving SIMPONI IV + MTX as compared with placebo IV + MTX (62.8% compared with 31.5%, respectively) (Table 2). (See Figure 1.)
Click on icon to see table/diagram/image
The percentage of patients achieving ACR 50 and ACR 70 responses was also greater in the SIMPONI IV + MTX group than in the placebo IV + MTX group (Table 4). The percentage of patients achieving an ACR 50 response for the SIMPONI IV + MTX and placebo IV + MTX groups, was 29.9% and 8.6%, respectively, at Week 14 (p<0.001), and 34.9% and 13.2%, respectively, at Week 24 (p<0.001). The percentage of patients achieving an ACR 70 response for the SIMPONI IV + MTX and placebo IV + MTX groups was 12.4% and 3.0%, respectively, at Week 14 (p<0.001) and 17.5% and 4.1%, respectively at Week 24 (p<0.001).
The proportions of patients achieving an ACR 20, ACR 50, or ACR 70 response were maintained after Week 24 through Week 52.
The percentage of patients achieving a DAS28 (using CRP) response was significantly greater for those patients treated with SIMPONI IV + MTX compared with those who received placebo IV + MTX at Week 14 (81.3% compared with 40.1%; p<0.001) and at Week 24 (81.0% compared with 44.7%; p<0.001). The percentage of patients achieving DAS28 < 2.6 (using CRP) remission was significantly greater for those patients treated with SIMPONI IV + MTX compared with those who received placebo IV + MTX at Week 14 (15.4% compared with 4.6%; p<0.001) and at Week 24 (17.7% compared with 5.1%; p<0.001). Of patients treated with SIMPONI + MTX who achieved DAS28 remission, 43% had no active joints, 27% had one active joint, 23% had two active joints, and 7% had three or more active joints, where an active joint was a joint that was rated as tender or swollen or both. The proportions of SIMPONI -treated patients achieving a DAS28 response or remission (using CRP) at Week 24 were maintained through Week 52. (See Table 2.)
Click on icon to see table/diagram/image
SIMPONI IV + MTX treatment also resulted in significantly greater improvement for each ACR component compared with treatment with placebo + MTX (Table 3). Swollen joint count for the SIMPONI IV + MTX and placebo IV + MTX group improved by 68% and 25%, respectively, at Week 14 and 75% and 27%, respectively, at Week 24. Improvement in tender joint count was 62% compared with 13% at Week 14, and 64% compared with 14% at Week 24, for the SIMPONI IV + MTX and placebo IV + MTX group, respectively. Patients' and physicians' assessments and HAQ score also were significantly improved for SIMPONI IV + MTX compared with placebo IV + MTX at Week 14 and Week 24. For SIMPONI IV + MTX, there was a 78% improvement in CRP compared with 29% improvement for placebo + MTX at Week 14, and 77% compared with 21% at Week 24. The improvement in all ACR components observed at Week 24 was maintained at each visit through Week 52. (See Table 3.)
Click on icon to see table/diagram/image
Radiographic response: In GO-BEFORE the change from baseline in the vdH-S score, a composite score of structural damage that radiographically measures the number and size of joint erosions and the degree of joint space narrowing in hands/wrists and feet, was used to assess the degree of structural damage. Key results for the SIMPONI 50 mg dose at week 52 are presented in Table 4.
The number of patients with no new erosions or a change from baseline in total vdH-S Score ≤ 0 was significantly higher in the SIMPONI treatment group than in the control group (p = 0.003). The radiographic effects observed at week 52 were maintained through week 104. Among patients remaining in the study and treated with SIMPONI, radiographic effects were similar from week 104 through week 256. (See Table 4.)
Click on icon to see table/diagram/image
In IV RA Study 1, structural joint damage was assessed radiographically and expressed as a change in van der Heijde-Modified Sharp Score (vdH-S), at Week 24 compared to baseline. The SIMPONI IV + MTX treatment group significantly inhibited the progression of structural damage compared with placebo + MTX, as assessed by total vdH-S score as shown in Table 5. Inhibition of radiographic progression continued to be observed in patients receiving SIMPONI IV + MTX at Week 52. (See Table 5.)
Click on icon to see table/diagram/image
At Week 24, a significantly greater proportion of patients in the SIMPONI IV + MTX group (71%) had no progression of structural damage (change in the total vdH-S score ≤ 0), compared to 57% of patients in the placebo + MTX group (Table 6). (See Table 6.)
Click on icon to see table/diagram/image
Physical function and health-related quality of life: Physical function and disability were assessed as a separate endpoint in GO-FORWARD and GO-AFTER using the disability index of the HAQ. In these studies, SIMPONI demonstrated clinically meaningful and statistically significant improvement in HAQ DI from baseline versus control at week 24. Among patients who remained on the SIMPONI treatment to which they were randomised at study start, improvement in HAQ DI was maintained through week 104. Among patients remaining in the study and treated with SIMPONI, improvement in HAQ DI was similar from week 104 through week 256.
In GO-FORWARD clinically meaningful and statistically significant improvements were demonstrated in health-related quality of life as measured by the physical component score of the SF-36 in patients treated with SIMPONI versus placebo at week 24. Among patients who remained on the SIMPONI treatment to which they were randomised at study start, improvement of the SF-36 physical component was maintained through week 104. Among patients remaining in the study and treated with SIMPONI, improvement of the SF-36 physical component was similar from week 104 through week 256. In GO-FORWARD and GO-AFTER, statistically significant improvements were observed in fatigue as measured by functional assessment of chronic illness therapy-fatigue scale (FACIT-F).
In IV RA Study 1, physical function and health-related quality of life were assessed using the disability index of the HAQ, the SF-36 health survey, and the Functional Assessment of Chronic Illness Therapy - Fatigue scale (FACIT-F). Patients treated with SIMPONI IV + MTX showed significantly greater median improvements in the HAQ compared with placebo IV + MTX at Week 14 (median 0.50 vs. 0.12; p<0.001) and Week 24 (median 0.50 vs. 0.12; p<0.001). The mean (± SD) improvement in the HAQ from baseline to Week 14 was 0.50 ± 0.58 for the SIMPONI IV + MTX group and 0.19 ± 0.56 for the placebo IV + MTX group. The mean improvement from baseline to Week 24 was 0.53 ± 0.64 for the SIMPONI IV + MTX group and 0.20 ± 0.55 for the placebo group (see Table 7). The mean improvement in HAQ was maintained through Week 52. The proportion of patients achieving a clinically meaningful ≥ 0.25 improvement in HAQ from baseline at Week 14 was greater in the SIMPONI IV + MTX group than in the placebo IV + MTX group (68% compared with 43%; p<0.001). This response was maintained at Week 24 (67% in the SIMPONI IV + MTX group vs. 45% in the placebo IV + MTX group; p<0.001) and through Week 52. (See Table 7.)
Click on icon to see table/diagram/image
In IV RA Study 1, median changes from baseline in both the SF-36 physical component summary scores and mental component summary scores were significantly greater with SIMPONI IV + MTX compared with patients treated with placebo IV + MTX at Week 12, Week 16, and Week 24 (p<0.001 for each component at all time points). Improvements in SF-36 physical component scores and mental component scores were maintained through Week 52.
SIMPONI IV + MTX showed significantly greater median improvement from baseline in the SF-36 physical component summary (PCS) score compared with placebo IV + MTX at Week 12 (5.6 vs. 2.6; p<0.001), Week 16 (7.1 vs. 1.9; p<0.001) and Week 24 (7.4 vs. 3.3; p<0.001). At baseline, the mean SF-36 PCS scores of 30.8 ± 6.8 in the SIMPONI IV + MTX group and 30.9 ± 7.3 in the placebo IV + MTX group were lower than the norm of 50 ± 10 measured in the US population. The mean improvement from baseline in the PCS score of the SF-36 at Week 12 was greater in the SIMPONI IV + MTX group than in the placebo IV + MTX group (5.9 ± 7.7 vs. 3.2 ± 7.4, respectively). The mean improvement on the SF-36 PCS score was maintained through Week 16, Week 24, and Week 52.
SIMPONI IV + MTX showed significantly greater median improvement from baseline in the SF-36 mental component summary (MCS) score compared with placebo IV + MTX at Week 12 (4.3 vs. 0.6; p<0.001), Week 16 (6.4 vs. 1.9; p<0.001) and Week 24 (6.4 vs. 1.1; p<0.001). The mean change from baseline in the MCS score of the SF-36 at Week 12 was greater in the SIMPONI IV + MTX group than in the placebo IV + MTX group (4.9 ± 10.3 vs. 1.5 ± 9.9, respectively). The mean improvement on the SF-36 MCS score was maintained through Week 16, Week 24, and Week 52.
In addition, the eight scales of the SF-36 that comprise the PCS and MCS scores showed greater median improvements at Week 12, Week 16, and Week 24 for the SIMPONI IV + MTX group compared to the placebo IV + MTX group (p<0.001 for all weeks and all scales). Compared to Week 24, improvements in each of the eight SF-36 scales were all maintained at Week 52.
Patients treated with SIMPONI IV + MTX had a clinically significant median improvement in fatigue as measured by FACIT-F. The median change from baseline in the FACIT-F score was significantly greater for SIMPONI IV + MTX compared with the placebo IV + MTX group at Week 16 (7.0 vs. 2.0; p<0.001) and Week 24 (8.0 vs. 2.0; p<0.001). Improvements in FACIT-F observed at Week 24 were maintained at Week 52. The median change from baseline in FACIT-F through Week 16 and Week 24 in the SIMPONI IV + MTX group was ≥ 4 points, which indicated a clinically meaningful improvement on fatigue. The mean change from baseline in the FACIT-F scores was greater in the SIMPONI IV + MTX group than in the placebo IV + MTX group at Week 16 (7.54 ± 10.546 vs. 2.16 ± 9.700, respectively). The mean improvement in the FACIT-F score was maintained through Week 24 in the SIMPONI IV + MTX group when compared with placebo (7.96 ± 10.793 vs. 2.54 ± 10.219 respectively, p<0.001). In addition, the proportion of patients achieving a clinically meaningful improvement (≥ 4 points) for FACIT-F at Week 24 was maintained at Week 52.
Health economics: Health economics data on health care resource utilization, time lost from work by patients and caregivers, employability, and patient productivity at work, school, or home were collected through questionnaires at baseline and every 8 weeks thereafter. Patients were asked to indicate how much their disease affected their productivity in the previous 4 weeks using a 0 to 10 cm VAS scale (not at all affected to affected very much). Only those assessments where statistically significant differences were observed between SIMPONI ± MTX and placebo ± MTX are presented as follows.
IV RA study 1: The median change from baseline in health state VAS scale (EQ VAS) was significantly greater in the SIMPONI IV + MTX group than in the placebo IV + MTX group at Week 16 and Week 24. At Week 24, the median improvement from baseline in EQ VAS scale was 20.00 in the SIMPONI + MTX group compared with 6.00 in the placebo + MTX group (p<0.001), and exceeded the threshold of clinically meaningful change defined as ½ standard deviation of baseline value (24.86/2=12.5).
Median decrease in self-reported impact of disease on productivity was significantly greater in the SIMPONI IV + MTX group compared with the placebo IV + MTX group at Week 24 (-2.9 vs. -0.5; p<0.001). The mean ± SD decrease in self-reported impact of disease on productivity in the SIMPONI IV + MTX group and placebo IV + MTX group at Week 24 was -2.78 ± 2.92 vs. -1.03 ± 2.96, respectively (p<0.001).
Psoriatic arthritis: The safety and efficacy of SIMPONI were evaluated in a multi-centre, randomized, double-blind, placebo-controlled study (GO-REVEAL) in 405 adult patients with active PsA (≥ 3 swollen joints and ≥ 3 tender joints) despite non-steroidal anti-inflammatory (NSAID) or DMARD therapy. Patients in this study had a diagnosis of PsA for at least 6 months and had at least mild psoriatic disease. Patients with each sub-type of psoriatic arthritis were enrolled, including polyarticular arthritis with no rheumatoid nodules (43%), asymmetric peripheral arthritis (30%), distal interphalangeal (DIP) joint arthritis (15%), spondylitis with peripheral arthritis (11%), and arthritis mutilans (1%). Previous treatment with an anti-TNF agent was not allowed. SIMPONI or placebo were administered subcutaneously every 4 weeks. Patients were randomly assigned to placebo, SIMPONI 50 mg, or SIMPONI 100 mg. Patients receiving placebo were switched to SIMPONI 50 mg after week 24. Patients entered an open label long-term extension at week 52. Approximately forty-eight percent of patients continued on stable doses of methotrexate (≤ 25 mg/week). The co-primary endpoints were the percentage of patients achieving ACR 20 response at week 14 and change from baseline in total PsA modified vdH-S score at week 24.
In general, no clinically meaningful differences in measures of efficacy were observed between the SIMPONI 50 mg and 100 mg dosing regimens through week 104. By study design, patients in the long-term extension may have switched between the 50 mg and 100 mg SIMPONI doses at the discretion of the study physician.
IV PsA study: The efficacy and safety of SIMPONI IV were evaluated in a multicenter, randomized, double-blind, placebo-controlled trial (GO-VIBRANT) in 480 adults with active psoriatic arthritis despite nonsteroidal anti-inflammatory drug (NSAID) or disease-modifying antirheumatic drug (DMARD) therapy. Patients in this trial had a diagnosis of PsA for at least six months and had symptoms of active disease (≥5 swollen joints, ≥5 tender joints, and a CRP level of ≥0.6 mg/dL). Patients were randomized to receive SIMPONI IV 2 mg/kg (N=241) or placebo (N=239) as a 30-minute intravenous infusion at Weeks 0, 4, 12 and 20. All patients on placebo received SIMPONI IV at Week 24, Week 28 and every 8 weeks thereafter through Week 52. Patients in the group treated with SIMPONI IV continued to receive infusions of SIMPONI IV at Week 28 and every 8 weeks through Week 52. Previous treatment with a biologic was not allowed.
Patients were allowed to continue stable doses of MTX, NSAIDs, and low dose oral corticosteroids (equivalent to ≤ 10 mg of prednisone per day) during the study. At study enrollment, the use of other DMARDs including cytotoxic agents or other biologics was prohibited.
Patients with each subtype of PsA were enrolled, including polyarticular arthritis with absence of rheumatoid nodules (44%), asymmetric peripheral arthritis (19%), distal interphalangeal joint involvement (8.1%), spondylitis with peripheral arthritis (25%), and arthritis mutilans (4.8%). The median duration of PsA disease was 3.5 years,86% of patients had previously used MTX, and 35% of patients received at least one other DMARD in the past. At baseline, 76% and 54% of the patients had enthesitis and dactylitis, respectively. During the trial, the use of concomitant medications was MTX (70%), oral corticosteroids (28%), and NSAIDs (71%).
The primary endpoint was the percentage of patients achieving an ACR 20 response at Week 14. The major secondary endpoints were change in baseline in the HAQ-DI score at Week 14, the proportion of subjects who achieve an ACR 50 response at Week 14, the proportion of subjects (with baseline ≥3% Body Surface Area [BSA] psoriatic involvement) who achieve a PASI 75 response at Week 14, and the change from baseline in total modified vdH-S score at Week 24.
Signs and symptoms: Key results for the 50mg dose at weeks 14 and 24 are shown in Table 8 and described as follows. (See Table 8.)
Click on icon to see table/diagram/image
Responses were observed at the first assessment (week 4) after the initial SIMPONI administration. Similar ACR 20 responses at week 14 were observed in patients with polyarticular arthritis with no rheumatoid nodules and asymmetric peripheral arthritis PsA subtypes. The number of patients with other PsA subtypes was too small to allow meaningful assessment. Responses observed in the SIMPONI treated groups were similar in patients receiving and not receiving concomitant MTX. Among 146 patients randomised to SIMPONI 50 mg, 70 were still on this treatment at week 104. Of these 70 patients, 64, 46 and 31 patients had an ACR 20/50/70 response, respectively. Among patients remaining in the study and treated with SIMPONI, similar rates of ACR 20/50/70 response was observed from week 104 through week 256.
Statistically significant responses in DAS28 were also observed at weeks 14 and 24 (p < 0.05).
At week 24, improvements in parameters of peripheral activity characteristic of psoriatic arthritis (e.g. number of swollen joints, number of painful/tender joints, dactylitis and enthesitis) were seen in the SIMPONI-treated patients. SIMPONI treatment resulted in significant improvement in physical function as assessed by HAQ DI, as well as significant improvements in health-related quality of life as measured by the physical and mental component summary scores of the SF-36. Among patients who remained on the SIMPONI treatment to which they were randomised at study start, DAS28 and HAQ DI responses were maintained through week 104. Among patients remaining in the study and treated with SIMPONI, DAS28 and HAQ DI responses were similar from week 104 through week 256.
IV PsA study: Treatment with SIMPONI IV, compared with placebo, resulted in a significant improvement in signs and symptoms as demonstrated by the percentage of patients with an ACR 20 response at Week 14 (see Table 9). In the SIMPONI IV-treated group, 75% of patients achieved an ACR 20 response compared with 22% in the placebo group (p<0.001). This benefit was consistently observed when assessed in SIMPONI IV-treated patients across the five PsA subtypes. ACR 20 responses observed in the SIMPONI IV-treated groups were similar in patients who were receiving and not receiving concomitant MTX. In addition, the percentage of patients achieving an ACR 20 response at Week 24 was significantly greater for patients receiving SIMPONI IV (77%) compared with patients receiving placebo (24%) (p<0.001) (Table 9).
The percentage of patients achieving ACR 50, ACR 70 and ACR 90 was greater in patients treated with SIMPONI IV compared with placebo (Table 9). The percentage of patients achieving an ACR 50 response at Week 14 was 44% for patients treated with SIMPONI IV and 6.3% for patients treated with placebo (p<0.001). At Week 24, the percentage of patients achieving an ACR 50 response was 54% and 6.3% (p<0.001) for SIMPONI IV and placebo groups, respectively. The percentage of patients achieving an ACR 70 response at Week 14 was 25% for patients treated with SIMPONI IV and 2.1% for patients treated with placebo (p<0.001). At Week 24, the percentage of patients achieving an ACR 70 response was 33% and 3.3% (p<0.001) for SIMPONI IV and placebo groups, respectively. The percentage of patients achieving an ACR 90 response at Week 14 was 3.7% for patients treated with SIMPONI IV and 0.4% for patients treated with placebo (p=0.011). At Week 24, the percentage of patients achieving an ACR 90 response was 6.2% and 0% (p<0.001) for SIMPONI IV and placebo groups, respectively (Table 9). (See Table 9.)
Click on icon to see table/diagram/image
The percentage of patients achieving ACR 20 responses by visit through Week 24 for the IV PsA study is shown in Figure 2. After the initial administration, ACR 20 responses were observed in 46% of SIMPONI IV-treated patients at the first assessment (Week 2) compared with 7.5% of placebo-treated patients. Responses for ACR 50 and ACR 70 were also observed as early as the first assessment (Week 2). (See Figure 2.)
Click on icon to see table/diagram/image
Table 10 shows the median percent improvement in the components of the ACR response criteria for the SIMPONI IV and placebo groups in the IV PsA study. (See Table 10.)
Click on icon to see table/diagram/image
Table 11 shows the median change from baseline at Week 14 in the components of the ACR response criteria for the SIMPONI IV and placebo groups in the IV PsA study. (See Table 11.)
Click on icon to see table/diagram/image
The percentage of patients achieving modified Psoriatic Arthritis Response Criteria (mPsARC), Disease Activity Score including 28 joints using C-reactive protein (DAS28) response, and DAS28 remission was also greater in patients treated with SIMPONI IV compared with placebo at Week 14 and Week 24 (Table 12). The percentage of patients achieving an mPsARC response at Week 14 and Week 24 was 84% for patients treated with SIMPONI IV and 34% for patients treated with placebo (p<0.001).At baseline, the mean DAS28 score in the SIMPONI IV-treated group and the placebo-treated group was 5.4 and 5.5, respectively. The percentage of patients achieving a DAS28 response at Week 14 was 93% for patients treated with SIMPONI IV and 40% for patients treated with placebo (p<0.001). At Week 24, the percentage of patients achieving a DAS28 response was 93% and 40% (p<0.001) for SIMPONI IV and placebo groups, respectively. The percentage of patients achieving DAS28 remission at Week 14 was 37% for patients treated with SIMPONI IV and 4.6% for patients treated with placebo (p<0.001). At Week 24, the percentage of patients achieving DAS28 remission was 42% and 7.1% (p<0.001) for SIMPONI IV and placebo groups, respectively. The mean change from baseline in DAS28 score at Week 14 was -2.3 and -0.6 in patients treated with SIMPONI IV and placebo, respectively. At Week 24, the mean change from baseline in DAS28 score was -2.50 and -0.7 in patients treated with SIMPONI IV and placebo, respectively. A negative/decreasing DAS28 score is indicative of improvement. Responses for mPsARC and DAS28 were observed as early as the first assessment (Week 2). (See Table 12.)
Click on icon to see table/diagram/image
Table 13 shows the improvement in the enthesitis and dactylitis for the SIMPONI IV and placebo groups in the IV PsA study. Patients with enthesitis at baseline were evaluated for improvement using the Leeds Enthesitis Index (LEI) on a scale of 0-6. At baseline, the median LEI score in patients with enthesitis in the SIMPONI IV-treated group and the placebo-treated group was 3.0 and 3.0, respectively. SIMPONI IV-treated patients showed a median improvement in LEI score of 2.0 as compared with a median improvement in placebo-treated patients of 0.0 at Week 14 (p<0.001). Patients with dactylitis at baseline were evaluated for improvement on a scale of 0-60. At baseline, the median dactylitis score in patients with dactylitis in the SIMPONI IV-treated group and the placebo-treated group was 5.5 and 6.0, respectively. SIMPONI IV-treated patients showed a median improvement in dactylitis score of 4.0 compared with a median improvement of 2.0 in placebo-treated patients at Week 14 (p<0.001). Improvements in enthesitis and dactylitis were maintained in SIMPONI IV-treated group through Week 24. (See Table 13.)
Click on icon to see table/diagram/image
Patients are classified as achieving psoriatic arthritis minimal disease activity (MDA) if five of the following seven outcome measures are fulfilled: tender joint count ≤1; swollen joint count ≤1; PASI ≤1 or BSA ≤ 3; patient pain visual analog scale (VAS) score of ≤15; patient global disease activity VAS score of ≤20; HAQ score ≤0.5; and tender entheseal points ≤1. Greater proportions of patients in the SIMPONI IV-treated group compared with placebo achieved minimal disease activity responses at Weeks 14 (27% vs 4.2%) and 24 (34% vs 4.6%).
Radiographic response: Structural damage in both hands and feet was assessed radiographically by the change from baseline in the vdH-S score, modified for PsA by addition of hand distal interphalangeal (DIP) joints.
SIMPONI 50 mg treatment reduced the rate of progression of peripheral joint damage compared with placebo treatment at week 24 as measured by change from baseline in total modified vdH-S Score (mean ± SD score was 0.27 ± 1.3 in the placebo group compared with -0.16 ± 1.3 in the SIMPONI group; p = 0.011). Out of 146 patients who were randomized to SIMPONI 50 mg, 52-week X-ray data were available for 126 patients, of whom 77% showed no progression compared to baseline. At week 104, X-ray data were available for 114 patients, and 77% showed no progression from baseline. Among patients remaining in the study and treated with SIMPONI, similar rates of patients showed no progression from baseline from week 104 through week 256.
IV PsA study: Structural joint damage was assessed radiographically and expressed as a change in total modified van der Heijde-Sharp (vdH-S) score and its components, the erosion score and joint space narrowing (JSN) score, at Week 24 compared to baseline. At baseline, the mean total modified vdH-S score was 35.0, the mean erosion score was 21.7, and the mean JSN score was 13.3. No change from baseline or a decrease from baseline in total modified vdH-S, erosion, or JSN scores is indicative of the inhibition of progression. At Week 24, SIMPONI IV significantly inhibited the progression of structural damage compared with placebo, as assessed by total modified vdH-S score and its components as shown in Table 14. (See Table 14.)
Click on icon to see table/diagram/image
Among patients with ≥1 joint with a JSN score of 0 at baseline (92%), a greater proportion of SIMPONI IV-treated patients showed no new erosions and no new joint space narrowing compared with placebo-treated patients at Week 24 (see Table 15).
Click on icon to see table/diagram/image
At Week 24, a greater proportion of patients in the SIMPONI IV-treated group (72%) had no progression of structural damage (change in the total modified vdH-S score ≤ 0), compared to 43% of patients in the placebo group (p<0.001) (Table 16). (See Table 16.)
Click on icon to see table/diagram/image
Improvement in physical function and health-related quality of life: IV PsA Study: Physical function and health-related quality of life were assessed using the disability index of the HAQ (HAQ-DI), the SF-36 health survey, EQ 5D-5L (EuroQoL 5 Dimensions questionnaire) Index Score, EQ VAS Score, and the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-Fatigue) questionnaire.
Patients treated with SIMPONI IV showed significantly greater median improvement from baseline in the HAQ-DI score compared with placebo at Week 14 (0.63 vs. 0.13; p<0.001) and Week 24 (0.63 vs. 0.13; p<0.001). Improvement in physical function as assessed by HAQ-DI demonstrated that the proportion of patients who achieved clinically meaningful improvement of ≥ 0.3 in HAQ-DI score from baseline was greater in the SIMPONI IV-treated group compared to placebo at Week 14 (69% vs. 32%; p<0.001) and Week 24 (69% vs. 33%; p<0.001) (see Table 17).
Click on icon to see table/diagram/image
Patients treated with SIMPONI IV showed significantly greater median improvement from baseline in the SF-36 physical component summary (PCS) score compared with placebo at Week 14 (8.7 vs. 1.8; p<0.001) and Week 24 (9.3 vs. 1.5). At baseline, the median SF-36 PCS scores were 33 in the SIMPONI IV group and 34 for the placebo group. Greater proportions of subjects in the SIMPONI IV group compared with placebo achieved a clinically meaningful change from baseline in SF-36 PCS score (an increase of 5 or more units) from Week 8 (63% vs 26%) through Week 24 (70% vs 29%).
Patients treated with SIMPONI IV showed significantly greater median improvement from baseline in the SF-36 mental component summary (MCS) score compared with placebo at Week 14 (5.2 vs. 0.4; p<0.001) and Week 24 (4.1 vs. 1.3). At baseline, the median SF-36 MCS scores were 43 in the SIMPONI IV group and 42 for the placebo group. Greater proportions of subjects in the SIMPONI IV group compared with placebo achieved a clinically meaningful change from baseline in SF-36 MCS score (an increase of 5 or more units) from Week 8 (46% vs 27%) through Week 24 (47% vs 29%).
In addition, the eight domains of the SF-36 that comprise the PCS and MCS scores showed greater median improvements at Week 8, Week 14, and Week 24 for the SIMPONI IV group compared to the placebo group.
EuroQoL 5D-5L is a measure of health status to provide a measure of health for clinical and economic appraisal. EQ-5D-5L consists of 2 elements: The EQ-5D-5L descriptive system and the EQ visual analogue scale (EQ VAS). The EQ-5D descriptive system comprises the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort and anxiety/depression. EQ VAS records the respondent's self-rated health on a VAS, with the endpoints of "worst imaginable health" and "Best imaginable health".
Patients treated with SIMPONI IV achieved improvement from baseline in the median EQ-5D-5L Index Score and EQ-VAS Score compared to placebo-treated patients at Week 14 (0.14 vs. 0.02 and 20 mm vs. 4.0 mm, respectively). Median baseline values for EQ-5D-5L index score and EQ VAS score, which were comparable between placebo and SIMPONI IV arms for all subjects, were 0.6 and 48 mm, respectively (see Table 18).
Click on icon to see table/diagram/image
Functional Assessment of Chronic Illness Therapy - Fatigue (FACIT-F) questionnaire consists of 13 questions that assess a subject's level of fatigue and tiredness over the last 7 days on a scale from 0-52. At baseline, the median FACIT-Fatigue score in the SIMPONI IV-treated group and the placebo-treated group was 28. The median improvement from baseline in the FACIT-Fatigue score was significantly greater for SIMPONI IV compared with the placebo group at Week 14 (8.0 vs. 2.0; p<0.001) and Week 24 (8.0 vs. 3.0; p<0.001) (see Table 19). The median change from baseline in FACIT-Fatigue through Week 14 and Week 24 in the SIMPONI IV group was ≥ 4 points, which indicated a clinically meaningful improvement in fatigue. In addition, a greater proportion of SIMPONI IV-treated patients achieved a clinically meaningful improvement (≥ 4 points) from baseline for FACIT-Fatigue score compared with placebo at Weeks 14 and 24 (68% vs. 41% and 70% vs. 43%, respectively). (See Table 19.)
Click on icon to see table/diagram/image
Health economics: IV PsA Study: Health economics data on patient productivity at work was collected through the Work Limitations Questionnaire (WLQ) and productivity visual analog scale (VAS) at baseline and Weeks 8, 14 and 24.
The WLQ is a questionnaire to assess health-related work productivity loss. Patients who worked full or part time or volunteered were asked to rate their level of difficulty or ability to perform specific job demands to assess health-related work productivity loss through questionnaire on a scale of 0 (limited none of the time) to 100 (limited all of the time). Then the scale scores were converted into a Productivity Loss Score indicating an estimated percentage of at-work productivity loss due to health. At baseline, the median WLQ productivity loss score in the SIMPONI IV-treated group and the placebo-treated group was 8.8 and 8.9, respectively. The median decrease (improvement) from baseline in WLQ productivity loss score in the SIMPONI IV and placebo groups at Week 14 and 24 were -2.9 vs. -0.45 (p<0.001), and at Week 24 -3.2 vs. -0.7 (p<0.001).
Patients were asked to indicate how much their disease impacted their daily productivity using a 0 to 10 VAS scale (no impact on productivity (0) to high impact on productivity (10)). At baseline, the median impact of disease on daily productivity (VAS) in the SIMPONI IV -treated group and the placebo-treated group score was 6.4. The median decrease (improvement) from baseline in impact of disease on daily productivity (VAS) score in the SIMPONI IV and placebo groups at Week 14 and Week 24 was -3.4 vs. -0.45 (p<0.001) and -3.3 vs. -0.6 (p<0.001), respectively.
Axial spondyloarthritis: Ankylosing spondylitis: The safety and efficacy of SIMPONI were evaluated in a multi-centre, randomised, double-blind, placebo-controlled study (GO-RAISE) in 356 adult patients with active ankylosing spondylitis (defined as a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score ≥ 4 and a VAS for total back pain of ≥4, on a scale of 0 to 10 cm). Patients enrolled in this study had active disease despite current or previous NSAID or DMARD therapy and had not previously been treated with anti-TNF therapy. SIMPONI or placebo were administered subcutaneously every 4 weeks. Patients were randomly assigned to placebo, SIMPONI 50 mg and SIMPONI 100 mg and were allowed to continue concomitant DMARD therapy (MTX, SSZ and/or HCQ). The primary endpoint was the percentage of patients achieving Ankylosing Spondylitis Assessment Study Group (ASAS 20) response at week 14. Placebo-controlled efficacy data were collected and analysed through week 24.
Key results for the 50mg dose are shown in Table 20 and described as follows. In general, no clinically meaningful differences in measures of efficacy were observed between the SIMPONI 50 mg and 100mg dosing regimens through week 24. By study design, patients in the long-term extension may have switched between the 50 mg and 100 mg SIMPONI doses at the discretion of the study physician. (See Table 20.)
Click on icon to see table/diagram/image
Among patients remaining in the study and treated with SIMPONI, the proportion of patients with an ASAS 20 and ASAS 40 response were similar from week 24 through week 256.
Statistically significant responses in BASDAI 50, 70 and 90 (p ≤ 0.017) were also seen at weeks 14 and 24. Improvements in key measures of disease activity were observed at the first assessment (week 4) after the initial SIMPONI administration and were maintained through week 24. Among patients remaining in the study and treated with SIMPONI, similar rates of change from baseline in BASDAI were observed from week 24 through week 256. Consistent efficacy was seen in patients regardless of use of DMARDs (MTX, sulfasalazine and/or hydroxychloroquine), HLA-B27 antigen status or baseline CRP levels as assessed by ASAS 20 responses at week 14.
SIMPONI treatment resulted in significant improvements in physical function as assessed by changes from baseline in the BASFI at weeks 14 and 24. Health-related quality of life as measured by the physical component score of the SF-36 was also improved significantly at weeks 14 and 24. Among patients remaining in the study and treated with SIMPONI, improvements in physical function and health-related quality of life were similar from week 24 through week 256.
IV AS study: The efficacy and safety of SIMPONI IV were evaluated in a multicenter, randomized, double-blind, placebo-controlled trial (GO-ALIVE) in 208 adults with active ankylosing spondylitis and inadequate response or intolerance to NSAIDs. Patients had a diagnosis of definite AS for at least 3 months according to modified New York criteria. Patients had symptoms of active disease (Bath AS Disease Activity Index [BASDAI] ≥ 4, VAS for total back pain of ≥ 4, on scales of 0 to 10 cm (0 to 100 mm), and a CRP level of ≥ 0.3 mg/dL (3 mg/L)). Patients were randomized to receive SIMPONI IV 2 mg/kg (N=105) or placebo (N=103) as a 30-minute intravenous infusion at Weeks 0, 4 and 12. All patients on placebo received SIMPONI IV at Week 16, Week 20 and every 8 weeks thereafter through Week 52. Patients in the SIMPONI IV-treatment group continued to receive SIMPONI IV infusions at Week 20 and every 8 weeks through Week 52. Patients were allowed to continue stable doses of concomitant MTX, SSZ, hydroxychloroquine (HCQ), low dose oral corticosteroids (equivalent to ≤ 10 mg of prednisone per day), and/or NSAIDs during the trial. At study enrollment, the use of other DMARDs including cytotoxic agents or other biologics was prohibited.
The median duration of AS disease was 2.8 years, median duration of inflammatory back pain was 8 years, 90% were HLA-B27 positive, 8.2% had prior joint surgery or procedure, 5.8% had complete ankylosis of the spine, 14% had received prior therapy with one TNF blocker (other than golimumab) and discontinued for reasons other than lack of efficacy within the first 16 weeks of treatment (primary failure), and 76% received at least one DMARD in the past. During the trial, the use of concomitant medications was NSAIDs (88%), SSZ (38%), corticosteroids (26%), MTX (18%), and HCQ (0.5%).
The primary endpoint was the percentage of patients achieving an Assessment in Ankylosing Spondylitis (ASAS) 20 response at Week 16. The major secondary endpoints were the proportion of subjects who achieve an ASAS 40 response at Week 16, the proportion of subjects who achieve at least a 50% improvement from baseline in BASDAI at Week 16, and the change from baseline in BASFI at Week 16.
Reduction in signs and symptoms: IV AS study: Treatment with SIMPONI IV, compared with placebo, resulted in a significant improvement in signs and symptoms as demonstrated by the percentage of patients with an ASAS 20 response at Week 16 (see Table 21). In patients treated with SIMPONI IV, 73% of patients achieved ASAS 20 response compared with 26% treated with placebo (p<0.001) at Week 16. In addition, ASAS 40 response was achieved at Week 16 by more patients treated with SIMPONI IV (48%) compared with the placebo group (8.7%; p<0.001). At Week 16, a greater percentage of patients treated with SIMPONI IV achieved a low level of disease activity (i.e., ASAS partial remission, defined as a value < 2 on a scale of 0-10 cm in each of the four ASAS 20 response parameters) compared with patients treated with placebo (16% vs. 3.9%, p=0.003). Compared with placebo, patients treated with TRADENAME IV achieved significantly higher responses as measured by ASAS 5/6 response (65% vs. 12%, respectively, p<0.001). (See Table 21.)
Click on icon to see table/diagram/image
The percentage of patients achieving ASAS 20 responses by visit through Week 16 for IV AS Study is shown in Figure 3. ASAS 20 responses were observed in 37% of patients treated with SIMPONI IV at the first assessment (Week 2) compared with 19% placebo-treated patients. (See Figure 3.)
Click on icon to see table/diagram/image
Table 22 shows the improvements in the components of the ASAS response criteria and other measures of disease activity at Week 16 for the SIMPONI IV and placebo groups. (See Table 22.)
Click on icon to see table/diagram/image
BASDAI is a patient self-assessment using a visual analogue scale (0 to 10 cm) on the following criteria: fatigue, spinal pain, joint pain, enthesitis, qualitative morning stiffness, and quantitative morning stiffness. Patients treated with SIMPONI IV achieved significantly higher responses as measured by a 50%, 70% and 90% improvement from baseline in BASDAI at Week 16 (Table 23). The median baseline BASDAI score, assessed on a VAS scale (0-10 cm), was 7.1 for the SIMPONI IV-treated group and 7.0 for the placebo group. The median decrease (improvement) from baseline in BASDAI score was greater in SIMPONI IV-treated patients compared with placebo-treated patients at Week 16 (-3.0 vs. -0.7, respectively). The percentage of patients achieving a BASDAI 50 response at Week 16 was 41% for patients treated with SIMPONI IV and 15% for patients treated with placebo (p<0.001). At Week 16, a greater proportion of patients treated with SIMPONI IV achieved a BASDAI score of less than 3 compared to patients treated with placebo (38% vs. 13%, respectively). (See Table 23.)
Click on icon to see table/diagram/image
Patients with enthesitis at baseline (83%) were evaluated for improvement in enthesitis using the UCSF Enthesitis Index on a scale of 0-17. At baseline, the median UCSF Enthesitis Index score in the TRADENAME IV-treated group and the placebo-treated group was 5.0 and 6.0, respectively. TRADENAME IV-treated patients showed an improvement in enthesitis score of 3.0 as compared with an improvement in placebo-treated patients of 1.0 at Week 16 (p<0.001).
Ankylosing Spondylitis Disease Activity Score (ASDAS) is a measure of disease activity and is derived from assessments of total back pain, morning stiffness, patient global assessment, peripheral pain/swelling and measurement of CRP. Major improvement in ASDAS is defined as a decrease ≥ 2.0 and inactive disease defined as score of <1.3. In patients treated with SIMPONI IV, 52% of patients achieved ASDAS major improvement compared with 2.9% treated with placebo (p<0.001) at Week 16. At Week 16, a greater percentage of patients treated with SIMPONI IV achieved inactive disease compared with patients treated with placebo (20% vs. 2.9%, p<0.001).
Improvement in physical function: IV AS study: Bath Ankylosing Spondylitis Functional Index (BASFI) is average of 10 self-assessed questions related to patient's functional anatomy and ability to cope with everyday life. A decreasing score is indicative of improvement in BASFI. The median baseline BASFI score was 6.3 and 6.1 for the SIMPONI IV and placebo groups, respectively. Treatment with SIMPONI IV resulted in significant improvements in physical function as assessed by changes from baseline in BASFI over time and the percentage of patients who achieved a 2-unit improvement in BASFI at Week 16. At Weeks 2 and 16, the respective median decrease (improvement) from baseline in the BASFI for the golimumab and placebo groups were -0.9 and -0.3 at Week 2 and -2.2 vs -0.3 at Week 16. The improvement in physical function was maintained at Week 28 in patients treated with SIMPONI IV. Among patients with a BASFI score ≥ 2 at baseline, a greater percentage of patients treated with SIMPONI IV achieved a 2-unit improvement in BASFI compared with patients treated with placebo (59% vs. 22%, respectively) at Week 16 (see Table 24).
Click on icon to see table/diagram/image
Improvement in range of motion: IV AS study: Bath Ankylosing Spondylitis Metrology Index (BASMI) is a musculoskeletal assessment and is represented as an aggregate score of 5 components (lumbar flexion, lumbar side flexion, intermalleolar distance, tragus to wall distance, and cervical rotation). At Week 16, the median decrease (improvement) from baseline in BASMI was of greater magnitude in patients treated with SIMPNI IV (-0.4) compared to placebo (-0.05, p=0.001). The percentage of patients with at least 1-unit improvement in BASMI at Week 16 in patients treated with SIMPONI IV (14%) was greater compared with patients treated with placebo (5.0%, p=0.029) (see Table 25).
Click on icon to see table/diagram/image
Improvement in health-related quality of life: IV AS study: Health-related quality of life was measured by the SF 36 (Short Form 36 Health Survey) Physical and Mental Component Summaries, ASQoL (Ankylosing Spondylitis Quality of Life questionnaire), EQ 5D-5L (EuroQoL 5 Dimensions questionnaire) Index Score, and EQ VAS Score at baseline, Week 8, Week 16 and Week 28.
Patients receiving SIMPONI IV demonstrated significantly greater median improvement from baseline compared with placebo in the physical component summary (PCS, 7.8 vs. 2.7, p<0.001) score at Week 16. At baseline, the median SF-36 PCS scores of 31 in the SIMPONI IV group and 31 for the placebo group. Greater proportions of subjects in the SIMPONI IV group compared with placebo achieved a clinically meaningful change from baseline in SF-36 PCS score (an increase of 5 or more units) at Week 8 (58% vs 27%) and Week 16 (68% vs 36%).
Patients receiving SIMPONI IV demonstrated significantly greater median improvement from baseline compared with placebo in the mental component summary (MCS, 5.7 vs. 1.8, p<0.001) score at Week 16. At baseline, the median SF-36 MCS scores were 40 in the SIMPONI IV group and 41 for the placebo group. Greater proportions of subjects in the SIMPONI IV group compared with placebo achieved a clinically meaningful change from baseline in SF-36 MCS score (an increase of 5 or more units) from Week 8 (49% vs 34%) and Week 16 (54% vs 29%).
In addition, the eight domains of the SF-36 that comprise the PCS and MCS scores showed greater median improvements at Week 8 and Week 16 for patients treated with SIMPONI IV compared to the placebo.
Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL) is a disease-specific instrument used to measure quality of life in the AS patient population. It consists of 18 items requesting a yes or no response to questions related to the impact of pain on sleep, mood, motivation, ability to cope, activities of daily living, independence, relationships, and social life. Lower scores indicate improvement. Median baseline ASQoL scores in the SIMPONI IV-treated group and placebo group were 14 and 13, respectively. Patients treated with SIMPONI IV achieved greater median decrease (improvement) from baseline in the ASQoL score compared to placebo-treated patients at Week 16 (-4.0 vs. -1.0, p<0.001).
EuroQoL 5D-5L is a measure of health status to provide a measure of health for clinical and economic appraisal. EQ-5D-5L consists of 2 elements: The EQ-5D-5L descriptive system and the EQ visual analogue scale (EQ VAS). The EQ-5D descriptive system comprises the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort and anxiety/depression. EQ VAS records the respondent's self-rated health on a VAS, with the endpoints of "worst imaginable health" and "best imaginable health".
Patients treated with SIMPONI achieved median improvement from baseline in the EQ-5D-5L Index Score and EQ VAS Score compared to placebo-treated patients at Week 16 (0.2 vs. 0.05 and 20 mm vs. 5.0 mm, respectively). Median baseline values for EQ-5D-5L index score and EQ VAS score in the SIMPONI IV-treated group and placebo group were 0.5 vs. 0.6 and 37 mm vs. 37 mm, respectively (see Table 26).
Click on icon to see table/diagram/image
Health economics: IV AS study: Health economics data on patient productivity at work was collected through the Work Limitations Questionnaire (WLQ) and the productivity visual analog scale (VAS) at baseline and Weeks 8, 16 and 28.
The WLQ is a questionnaire to assess health-related work productivity loss. Patients who worked full or part time or volunteered were asked to rate their level of difficulty or ability to perform specific job demands to assess health-related work productivity loss through questionnaire on a scale of 0 (limited none of the time) to 100 (limited all of the time). Then the scale scores were converted in to a Productivity Loss Score indicating an estimated percentage of at-work productivity loss due to health. At baseline, the median WLQ productivity loss scores in the SIMPONI IV-treated group and placebo group were 11 and 11, respectively. The median decrease (improvement) from baseline in WLQ productivity loss score in the SIMPONI IV and placebo groups at Week 16 was -2.8 vs. -0.7.
Patients were asked to indicate how much their disease affected their productivity using a 0 to 10 VAS scale (no impact on productivity (0) to high impact on productivity (10)). At baseline, the median impact of disease on daily productivity (VAS) scores in the SIMPONI IV-treated group and placebo group were 7.6 and 7.3. The median decrease (improvement) from baseline in impact of disease on daily productivity (VAS) score in the SIMPONI IV and placebo groups at Week 16 was -2.6 vs. -0.7.
Improvement in sleep: IV AS study: In the IV AS study, the extent of sleep problems was assessed using the Medical Outcomes Study Sleep Scale (MOS-SS), which measured 6 dimensions of sleep, including sleep disturbance, somnolence, sleep adequacy, snoring, shortness of breath or headache, and quantity of sleep/optimal sleep. At baseline, the median sleep problems index score was 39 in the SIMPONI IV and placebo groups. At Week 16, median improvement in sleep problems index was greater for patients treated with SIMPONI IV compared with the placebo group (7.0 vs. 2.8). An increase from baseline represents improvement (Table 27).
Click on icon to see table/diagram/image
Non-radiographic axial spondyloarthritis: The safety and efficacy of SIMPONI were evaluated in a multi-centre, randomised, double-blind, placebo-controlled study (GO-AHEAD) in 197 adult patients with severe active nr-Axial SpA (defined as those patients meeting the ASAS classification criteria of axial spondyloarthritis but did not meet the modified New York criteria for AS). Patients enrolled in this study had active disease (defined as a BASDAI ≥ 4 and a Visual Analogue Scale (VAS) for total back pain of ≥ 4, each on a scale of 0-10 cm) despite current or previous NSAID therapy and had not previously been treated with any biological agents including anti-TNF therapy. Patients were randomly assigned to placebo or SIMPONI 50 mg administered subcutaneously every 4 weeks. At week 16, patients entered an open label period in which all patients received SIMPONI 50 mg administered subcutaneously every 4 weeks through week 48 with efficacy assessments performed through week 52 and safety follow-up through week 60.
Approximately 93% of patients who were receiving SIMPONI at the beginning of the open-label extension (week 16) remained on treatment through the end of the study (week 52). Analyses were performed on both the All Treated (AT, N = 197) and Objective Signs of Inflammation (OSI, N = 158, defined by elevated CRP and/or evidence of sacroiliitis on MRI at baseline) populations. Placebo-controlled efficacy data were collected and analysed through week 16. The primary endpoint was the proportion of patients achieving ASAS 20 response at week 16. Key results are shown in Table 28 and described as follows. (See Table 28.)
Click on icon to see table/diagram/image
Statistically significant improvements in signs and symptoms of severe active nr-Axial SpA were demonstrated in patients treated with SIMPONI 50 mg compared to placebo at week 16 (Table 28). Improvements were observed at the first assessment (week 4) after the initial SIMPONI administration. SPARCC score as measured by MRI showed statistically significant reductions in SI joint inflammation at week 16 in patients treated with SIMPONI 50 mg compared to placebo (Table 28). Pain as assessed by the Total Back Pain and Nocturnal Back Pain VAS, and disease activity as measured by ASDAS-C also showed statistically significant improvement from baseline to week 16 in patients treated with SIMPONI 50 mg compared to placebo (p < 0.0001).
Statistically significant improvements in spinal mobility as assessed by BASMI (Bath Ankylosing Spondylitis Metrology Index) and in physical function as assessed by the BASFI were demonstrated in SIMPONI 50 mg-treated patients as compared to placebo-treated patients (p < 0.0001). Patients treated with SIMPONI experienced significantly more improvements in health-related quality of life as assessed by ASQoL, EQ-5D, and physical and mental components of SF-36, and experienced significantly more improvements in productivity as assessed by greater reductions in overall work impairment and in activity impairment as assessed by the WPAI questionnaire than patients receiving placebo.
For all of the endpoints described above, statistically significant results were also demonstrated in the OSI population at week 16.
In both the AT and OSI populations, the improvements in signs and symptoms, spinal mobility, physical function, quality of life, and productivity observed at week 16 among patients treated with SIMPONI 50 mg continued in those remaining in the study at week 52.
Ulcerative colitis: The efficacy of SIMPONI was evaluated in two randomized, double-blind, placebo-controlled clinical studies in adult patients.
The induction study (PURSUIT-Induction) evaluated patients with moderately to severely active ulcerative colitis (Mayo score 6 to 12; Endoscopy subscore ≥ 2) who had an inadequate response to or failed to tolerate conventional therapies, or were corticosteroid dependent. In the dose confirming portion of the study, 761 patients were randomized to receive either 400 mg SIMPONI SC at week 0 and 200 mg at week 2, 200 mg SIMPONI SC at week 0 and 100 mg at week 2, or placebo SC at weeks 0 and 2. Concomitant stable doses of oral aminosalicylates, corticosteroids, and/or immunomodulatory agents were permitted. The efficacy of SIMPONI through week 6 was assessed in this study.
The results of the maintenance study (PURSUIT-Maintenance) were based on evaluation of 456 patients who achieved clinical response from previous induction with SIMPONI. Patients were randomized to receive SIMPONI 50 mg, SIMPONI 100 mg or placebo administered subcutaneously every 4 weeks. Concomitant stable doses of oral aminosalicylates, and/or immunomodulatory agents were permitted. Corticosteroids were to be tapered at the start of the maintenance study. The efficacy of SIMPONI through week 54 was assessed in this study. Patients who completed maintenance study through week 54 continued treatment in a study-extension, with efficacy evaluated through week 216. Efficacy evaluation in the study extension was based on changes in corticosteroid use, Physician's Global Assessment (PGA) of disease activity, and improvement in quality of life as measured by Inflammatory Bowel Disease Questionnaire (IBDQ). (See Table 29.)
Click on icon to see table/diagram/image
More SIMPONI-treated patients demonstrated sustained mucosal healing (patients with mucosal healing at both week 30 and week 54) in the 50mg group (42%, nominal p < 0.05) and 100 mg group (42%, p < 0.005) compared with patients in the placebo group (27%).
Among the 54% of patients (247/456) who were receiving concomitant corticosteroids at the start of PURSUIT-Maintenance, the proportion of patients who maintained clinical response through week 54 and were not receiving concomitant corticosteroids at week 54 was greater in the 50mg group (38%, 30/78) and 100 mg group (30%, 25/82) compared with the placebo group (21%, 18/87). The proportion of patients who eliminated corticosteroids by week 54 was greater in the 50mg group (41%, 32/78) and 100 mg group (33%, 27/82) compared with the placebo group (22%, 19/87). Among patients who entered the study extension, the proportion of subjects who remained corticosteroid free was generally maintained through week 216.
Patients who did not achieve clinical response at week 6 in the PURSUIT-Induction studies were dosed Simponi 100 mg every 4 weeks in the PURSUIT-Maintenance study. At week 14, 28% of these patients achieved response defined by partial Mayo score (decreased by ≥ 3 points compared with start of induction). At week 54, the clinical outcomes observed in these patients were similar to the clinical outcomes reported for the patients achieving clinical response at week 6.
At week 6, SIMPONI significantly improved quality of life as measured by change from baseline in a disease specific measure, IBDQ (inflammatory bowel disease questionnaire). Among patients who received SIMPONI maintenance treatment, the improvement in quality of life as measured by IBDQ was maintained through week 54.
Approximately 63% of patients who were receiving Simponi at the beginning of the study extension (week 56), remained on treatment through the end of the study (last golimumab administration at week 212).
Immunogenicity: Across the Phase III RA, PsA and AS studies through week 52, antibodies to golimumab were detected by the enzyme immunoassay (EIA) in 5% (105/2062) of golimumab-treated patients and, where tested, nearly all antibodies were neutralising in vitro. Similar rates were shown across rheumatologic indications. Treatment with concomitant MTX resulted in a lower proportion of patients with antibodies to golimumab than patients receiving golimumab without MTX (approximately 3% [41/1235] versus 8% [64/827], respectively).
In nr-Axial SpA, antibodies to golimumab were detected in 7% (14/193) of golimumab treated patients through week 52.
In the Phase II and III UC studies through week 54, antibodies to golimumab were detected by the EIA method in 3% (26/946) of golimumab treated patients. Sixty-eight percent (21/31) of antibody-positive patients had neutralizing antibodies in vitro. Treatment with concomitant immunomodulators (azathioprine, 6-mercaptopurine and MTX) resulted in a lower proportion of patients with antibodies to golimumab than patients receiving golimumab without immunomodulators (1% (4/308) versus 3% (22/638), respectively). Of patients that continued in the study extension and had evaluable samples through week 228, antibodies to golimumab were detected in 4% (23/604) of golimumab treated patients. Eighty-two percent (18/22) of antibody-positive patients had neutralizing antibodies
in vitro.
The presence of antibodies to golimumab may increase the risk of injection site reactions (see Precautions). The small number of patients positive for antibodies to golimumab limits the ability to draw definitive conclusions regarding the relationship between antibodies to golimumab and clinical efficacy or safety measures.
Because immunogenicity analyses are product- and assay-specific, comparison of antibody rates with those from other products is not appropriate.
Following IV administration of SIMPONI in combination with MTX in RA patients, antibodies to golimumab were detected in 4.2% (39/922) of golimumab-treated patients through approximately 1 year. All patients who were positive for antibodies to golimumab had neutralizing antibodies
in vitro.
The small number of patients positive for antibodies to SIMPONI limits the ability to draw definitive conclusions regarding the relationship between antibodies to golimumab and clinical efficacy or safety measures.
The data reflect the percentage of patients whose test results were considered positive for antibodies to SIMPONI in an ELISA assay, and are highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to SIMPONI with the incidence of antibodies to other products may be misleading.
Following IV administration in patients with RA or AS, antibodies to SIMPONI were detected by the drug-tolerant EIA method in 20% of SIMPONI-treated patients (RA: 21%, and AS: 19%). Where tested, approximately one-third of the antibodies to SIMPONI were neutralizing. Treatment with concomitant MTX resulted in a slightly lower proportion of patients with antibodies to SIMPONI than patients receiving SIMPONI without MTX (approximately 19% vs. 25%, respectively).
The higher incidence of antibodies to golimumab with the drug-tolerant EIA method were mostly due to low-titer antibodies, which did not have an apparent impact on drug concentrations, efficacy and safety. Although higher-titer antibodies, which were mostly neutralizing, may be associated with lower drug concentrations and diminished efficacy, there were few patients with high titers in the IV AS studies. Development of antibodies to SIMPONI did not preclude clinical response.
The observed incidence of antibody positivity may be influenced by several factors including sample handling, timing of sample collection, concomitant medications, underlying disease, and particularly the sensitivity of the assay. For these reasons, comparison of the incidence of antibodies to SIMPONI with the incidence of antibodies to other products or results from different assays may be misleading.
Pharmacokinetics: Absorption: Following a single subcutaneous administration of golimumab to healthy subjects or patients with RA, the median time to reach maximum serum concentrations (T
max) ranged from 2 to 6 days. A subcutaneous injection of 50 mg golimumab to healthy subjects produced a mean ± standard deviation maximum serum concentration (C
max) of 3.1 ± 1.4 mcg/ml.
Following a single subcutaneous injection of 100 mg, the absorption of golimumab was similar in the upper arm, abdomen, and thigh, with a mean absolute bioavailability of 51%. Since golimumab exhibited approximately dose proportional PK following a subcutaneous administration, the absolute bioavailability of a golimumab 50 mg or 200 mg dose is expected to be similar.
Distribution: Following a single IV administration, the mean volume of distribution was 115 ± 19 mL/kg.
Elimination: The systemic clearance of golimumab was estimated to be 6.9 ± 2.0 mL/day/kg. Terminal half-life value was estimated to be approximately 12 ± 3 days in healthy subjects and similar values were observed in patients with RA, PsA, AS, or UC.
When 50 mg golimumab was administered subcutaneously to patients with RA, PsA or AS every 4 weeks, serum concentrations reached steady state by Week 12. With concomitant use of MTX, treatment with 50 mg golimumab subcutaneously every 4 weeks resulted in a mean (± standard deviation) steady-state trough serum concentration of approximately 0.6 ± 0.4 mcg/ml in RA patients with active RA despite MTX therapy, and approximately 0.5 ± 0.4 mcg/ml in patients with active PsA and approximately 0.8 ± 0.4 mcg/ml in patients with AS. Steady-state trough mean serum golimumab concentrations in patients with nr-Axial SpA were similar to those observed in patients with AS following subcutaneous administration of 50 mg golimumab every 4 weeks.
Patients with RA, PsA or AS who did not receive concomitant MTX had approximately 30% lower steady-state trough concentrations of golimumab than those who received golimumab with MTX. In a limited number of RA patients treated with subcutaneous golimumab over a 6-month period, concomitant use of MTX reduced the apparent clearance of golimumab by approximately 36%. However, population pharmacokinetic analysis indicated that concomitant use of NSAIDs, oral corticosteroids or sulfasalazine did not influence the apparent clearance of golimumab.
Following induction doses of 200 mg and 100 mg golimumab at week 0 and 2, respectively, and maintenance doses of 50 mg or 100 mg golimumab subcutaneously every 4 weeks thereafter to patients with UC, serum golimumab concentrations reached steady state approximately 14 weeks after the start of therapy. Treatment with 50 mg or 100 mg golimumab subcutaneous every 4 weeks during maintenance resulted in a mean steady-state trough serum concentration of approximately 0.9 ± 0.5 mcg/mL and 1.8 ± 1.1 mcg/mL, respectively.
In UC patients treated with 50 mg or 100 mg golimumab subcutaneously every 4 weeks, concomitant use of immunomodulators did not have a substantial effect on steady-state trough levels of golimumab.
Patients who developed anti-golimumab antibodies generally had low trough steady-state serum concentrations of golimumab (see Pharmacodynamics previously).
Linearity: Golimumab exhibited approximately dose-proportional pharmacokinetics in patients with RA over the dose range of 0.1 to 10.0 mg/kg following a single intravenous dose. Following a single SC dose in healthy subjects, approximately dose-proportional pharmacokinetics were also observed over a dose range of 50 mg to 400 mg.
Effect of weight on pharmacokinetics: There was a trend toward higher apparent clearance of golimumab with increasing weight (see Dosage & Administration).
Toxicology: Preclinical Safety Data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, toxicity to reproduction and development.
No mutagenicity studies, animal fertility studies nor long-term carcinogenic studies have been conducted with golimumab.
In a fertility and general reproductive function study in mouse, using an analogous antibody that selectively inhibits the functional activity of mouse TNFα, the number of pregnant mice was reduced. It is not known whether this finding was due to effects on the males and/or the females. In a developmental toxicity study conducted in mice following administration of the same analogous antibody, and in cynomolgus monkeys using golimumab, there was no indication of maternal toxicity, embryotoxicity or teratogenicity.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image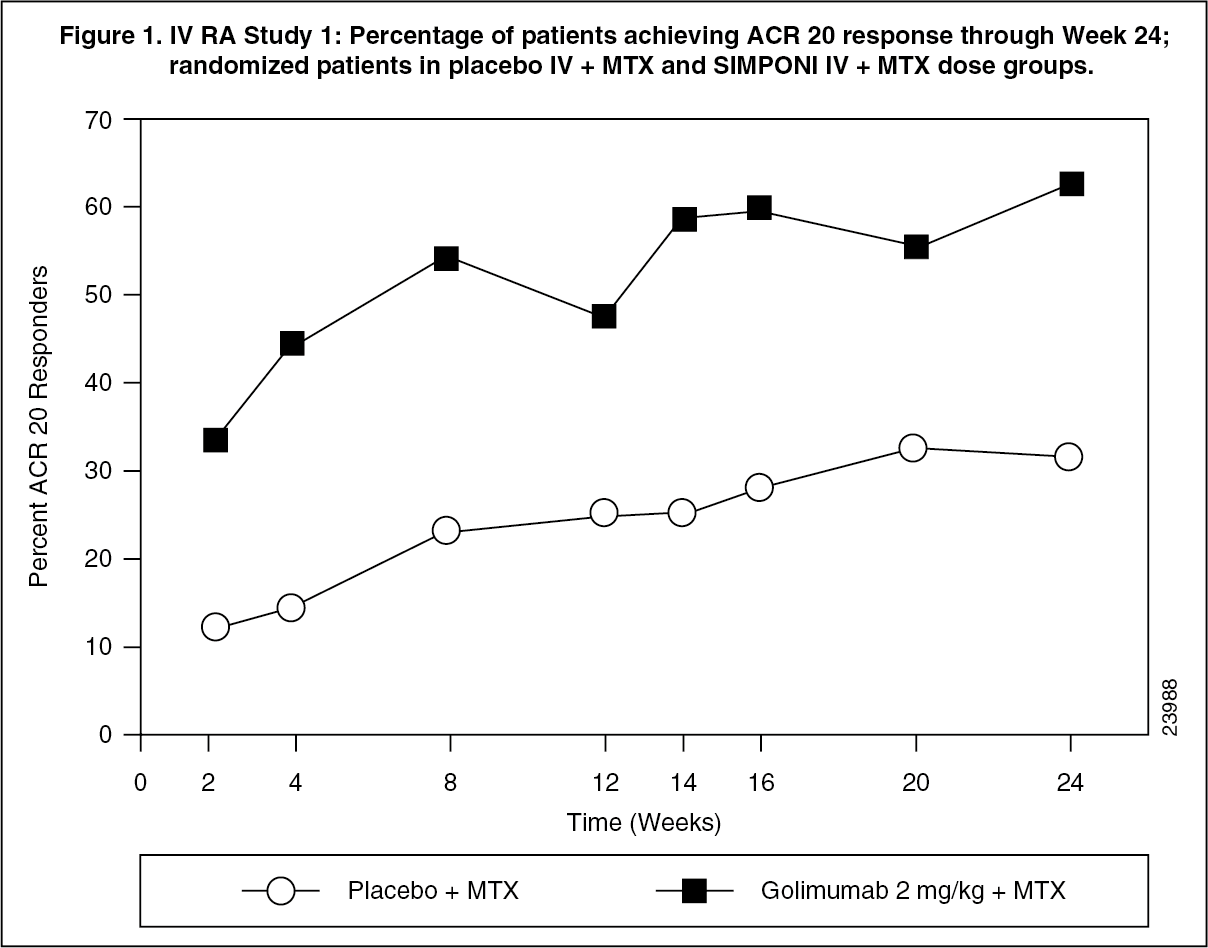 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image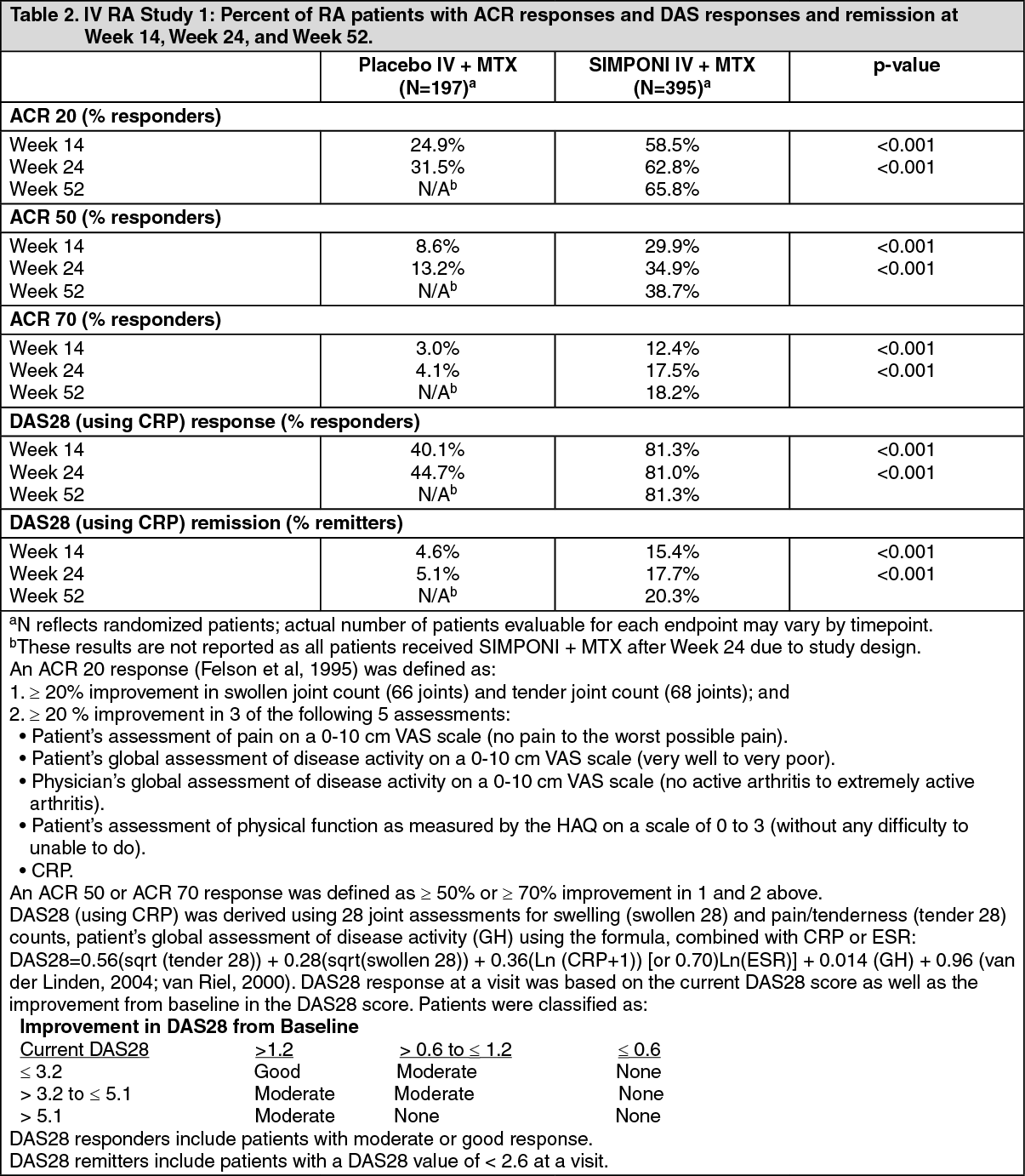 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image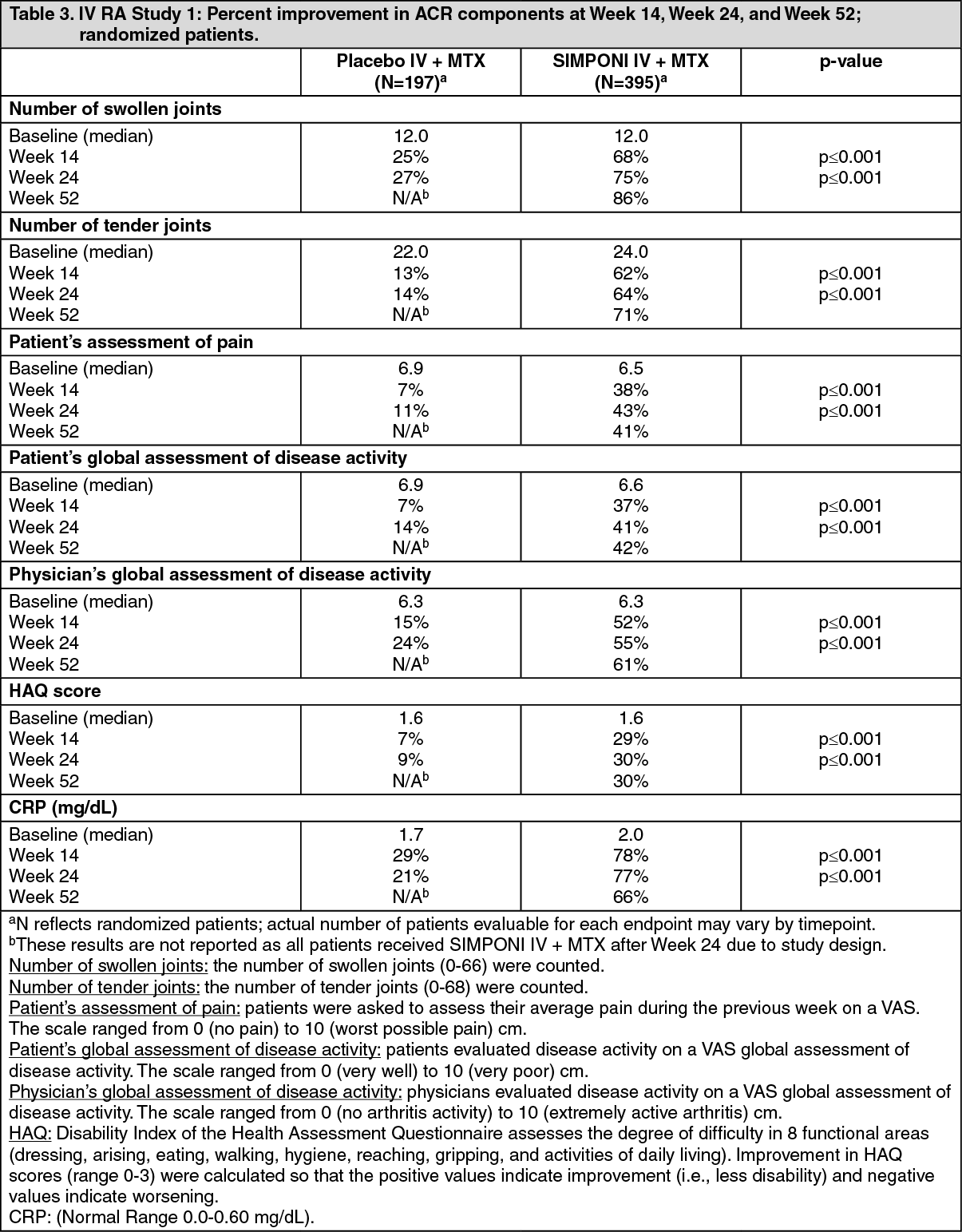 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image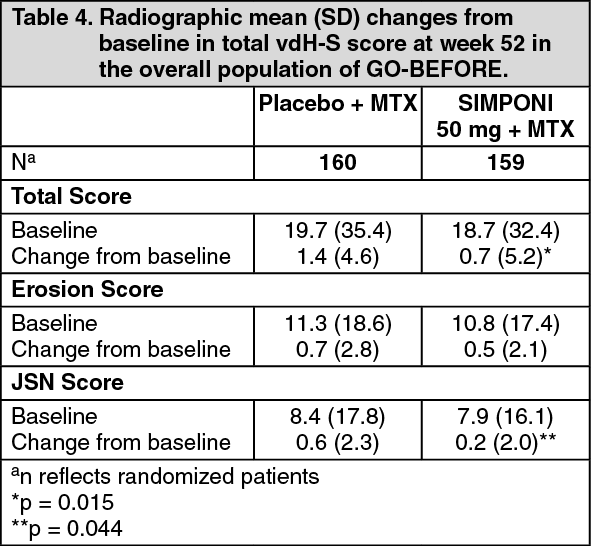 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image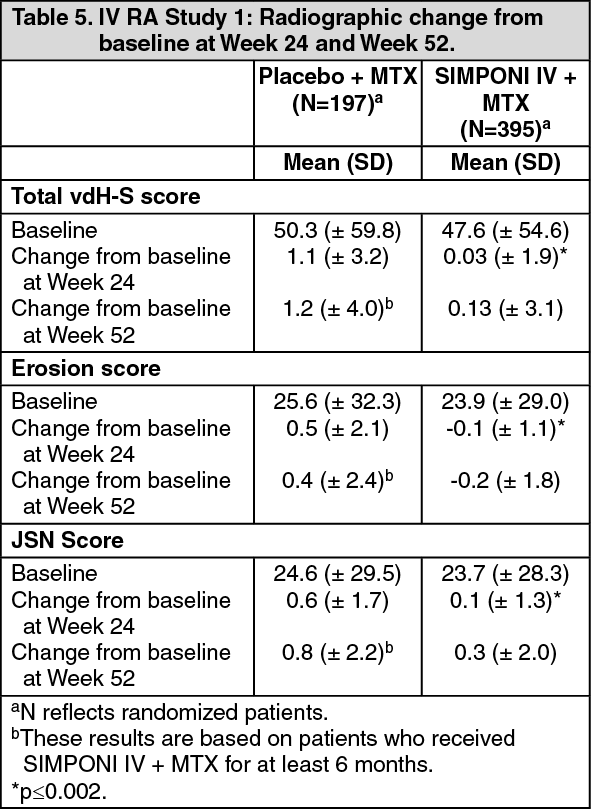 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image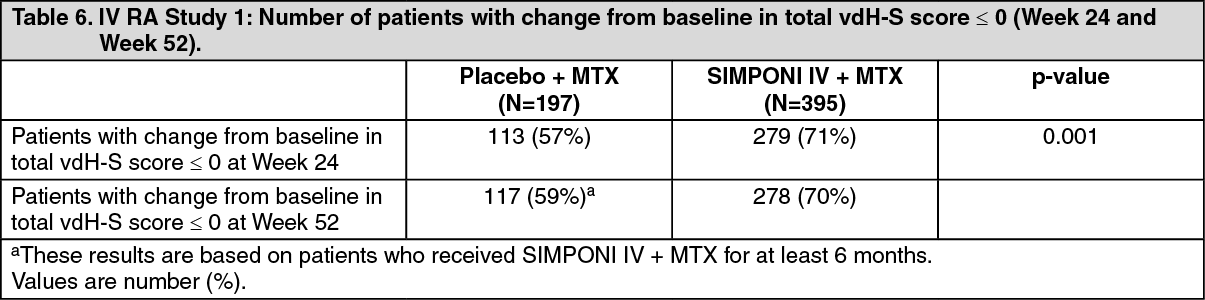 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image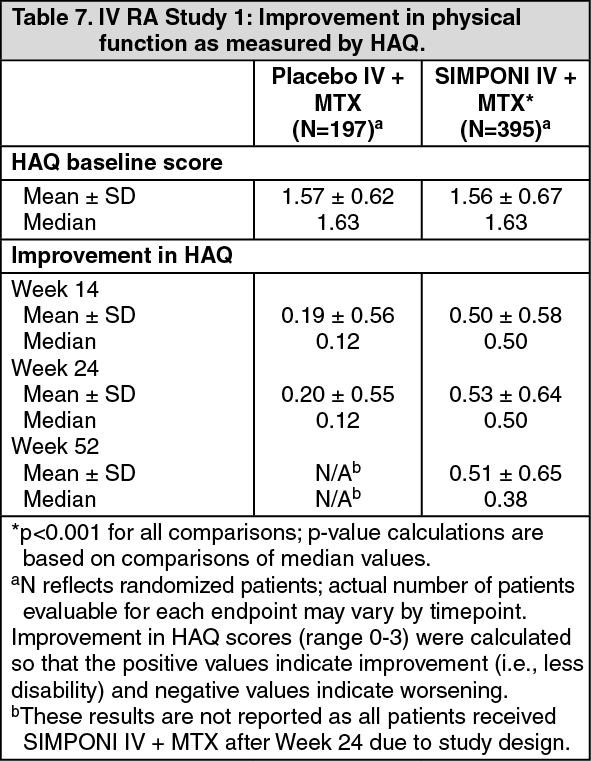 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image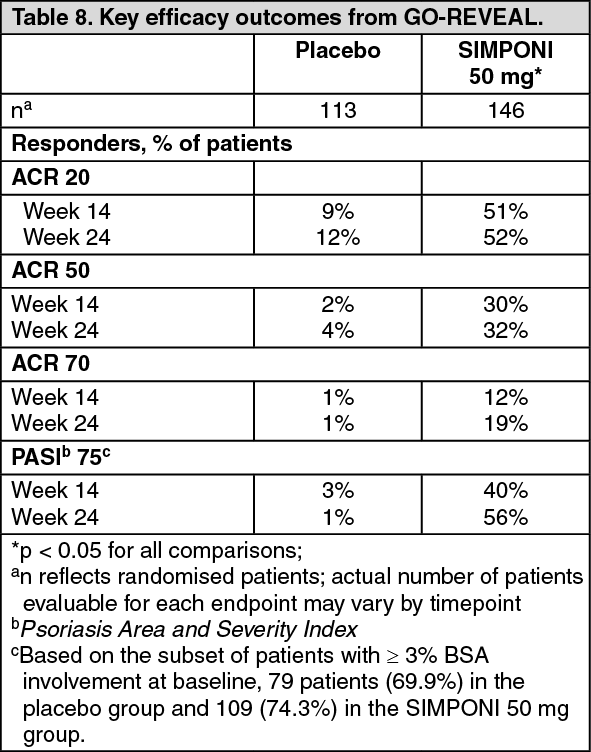 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image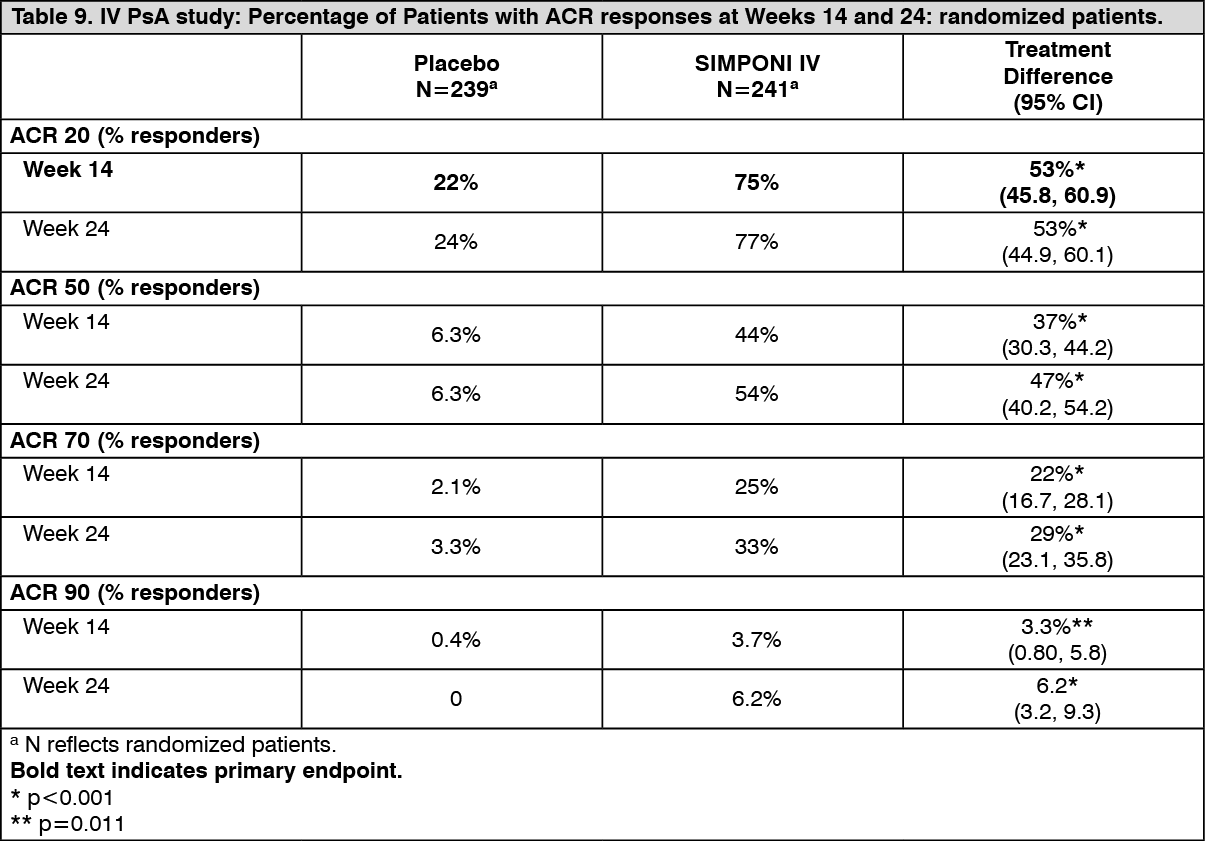 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image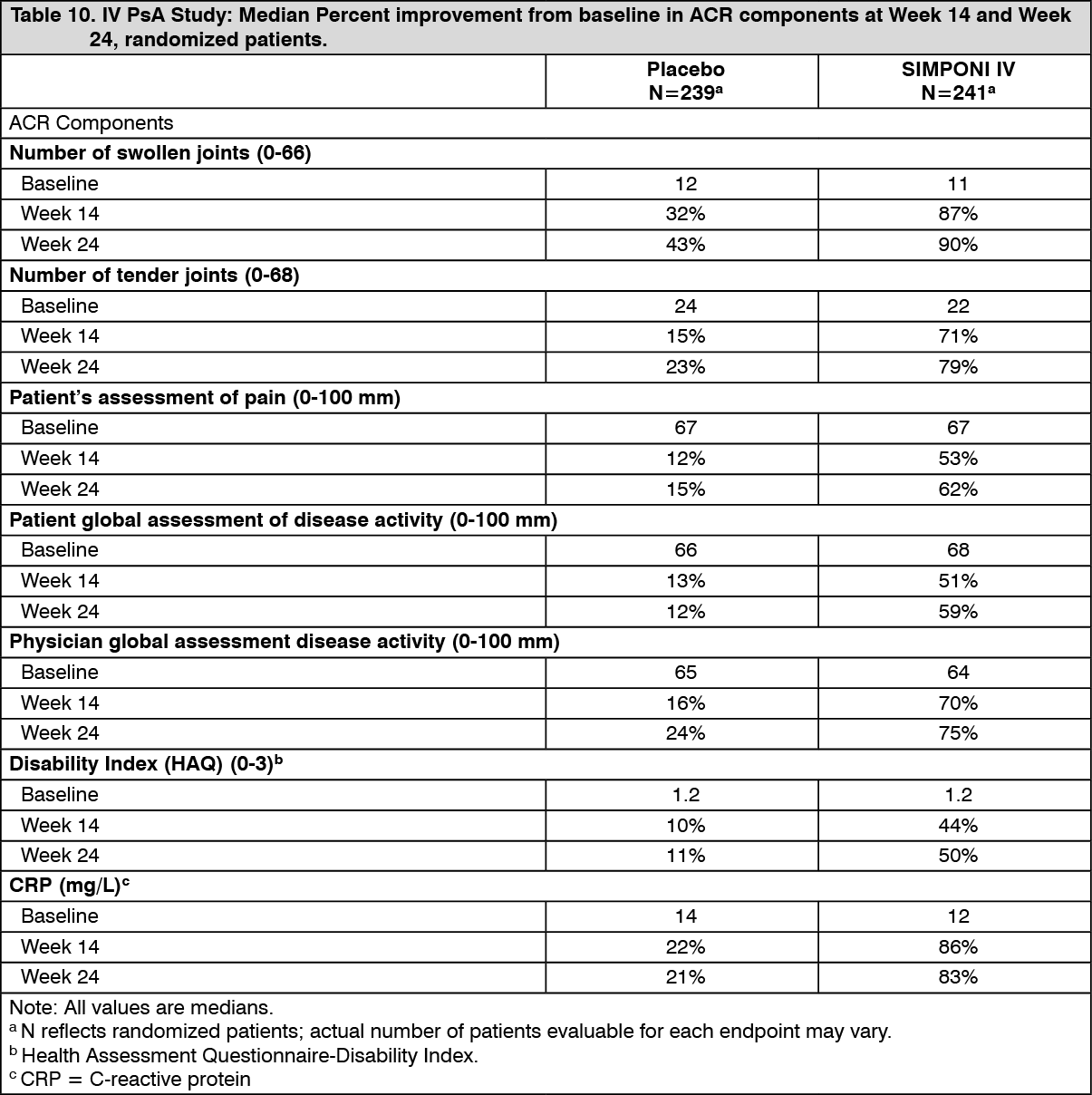 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image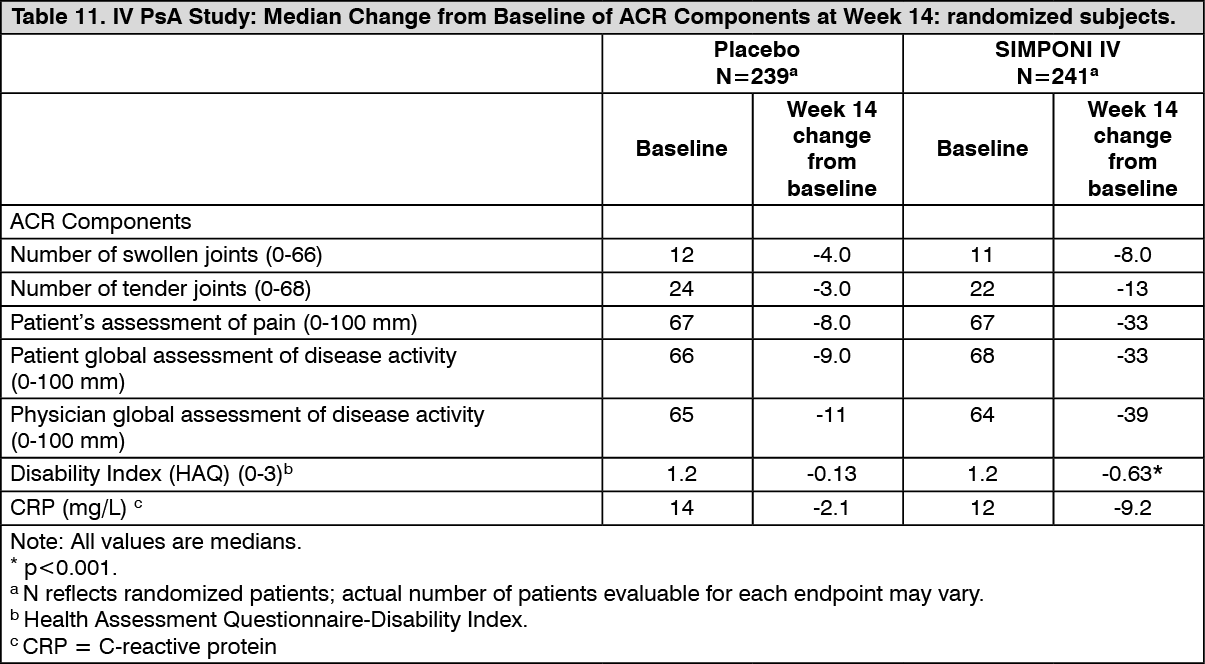 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image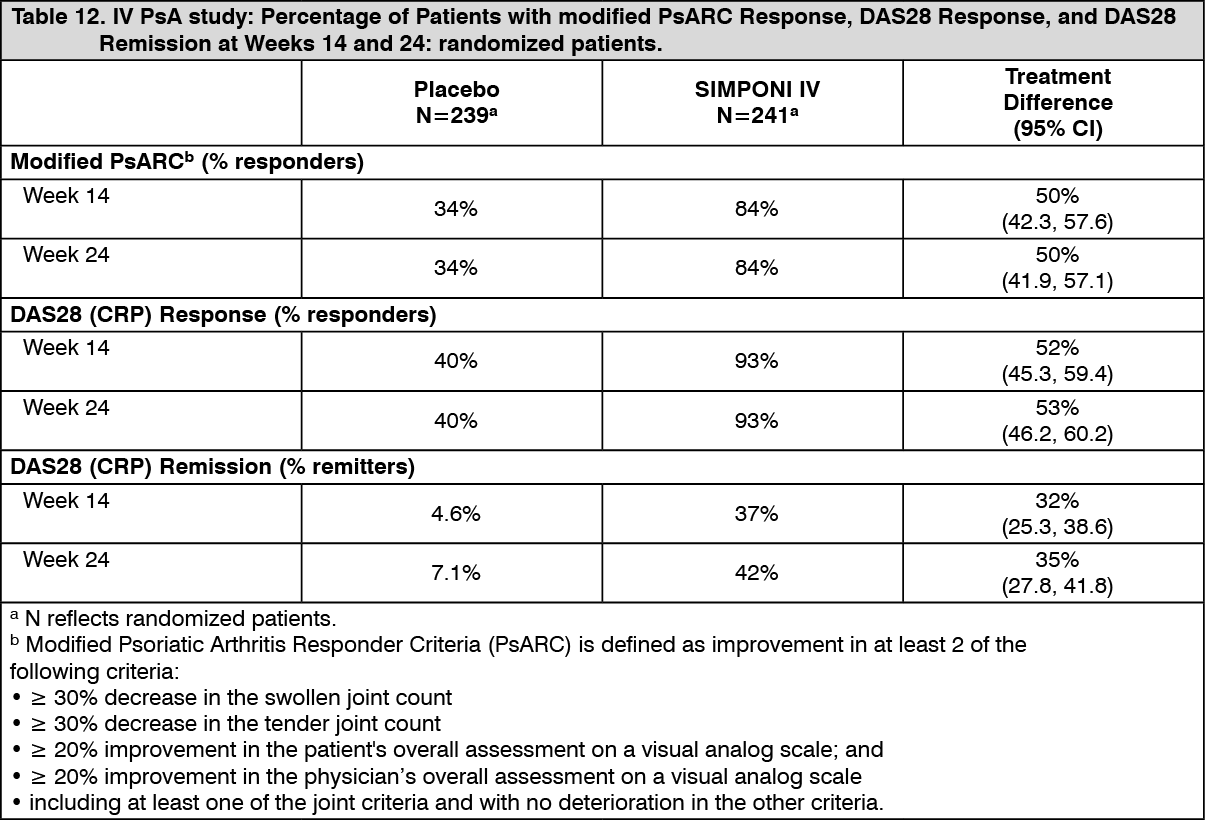 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image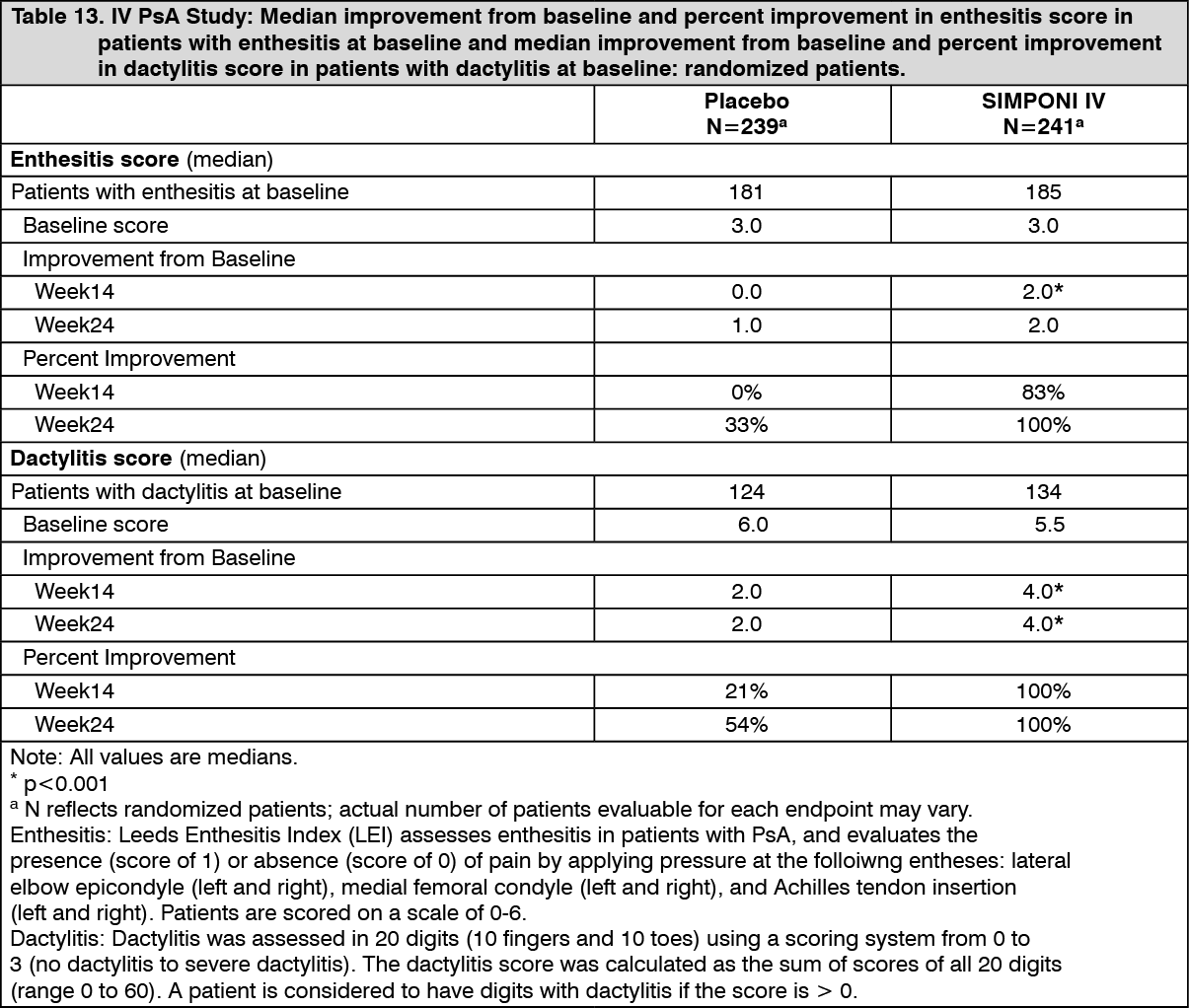 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image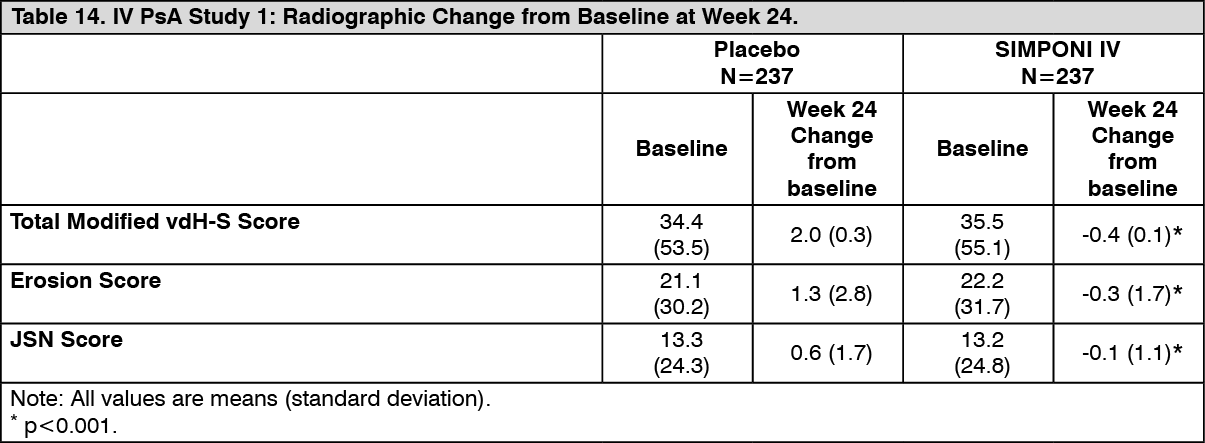 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image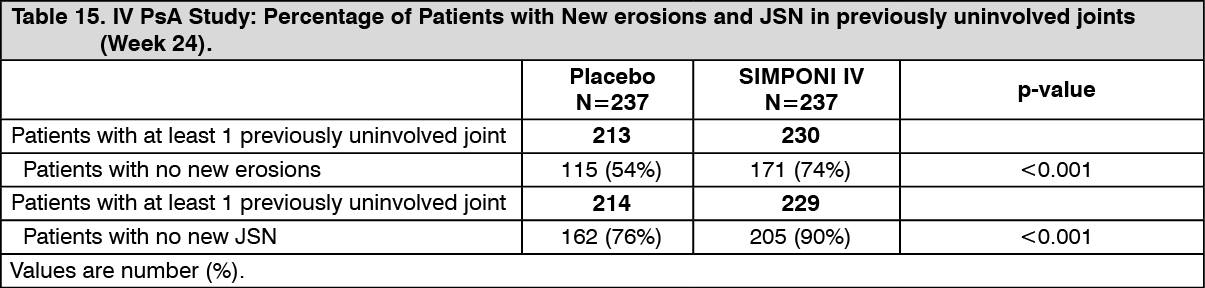 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image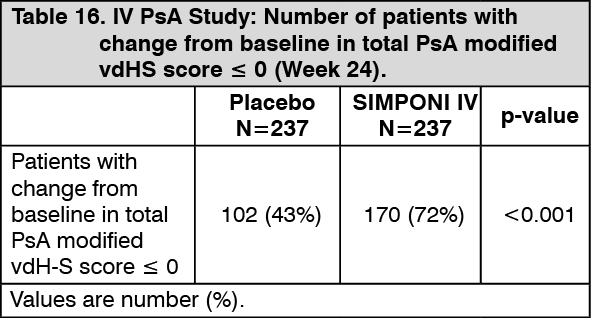 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image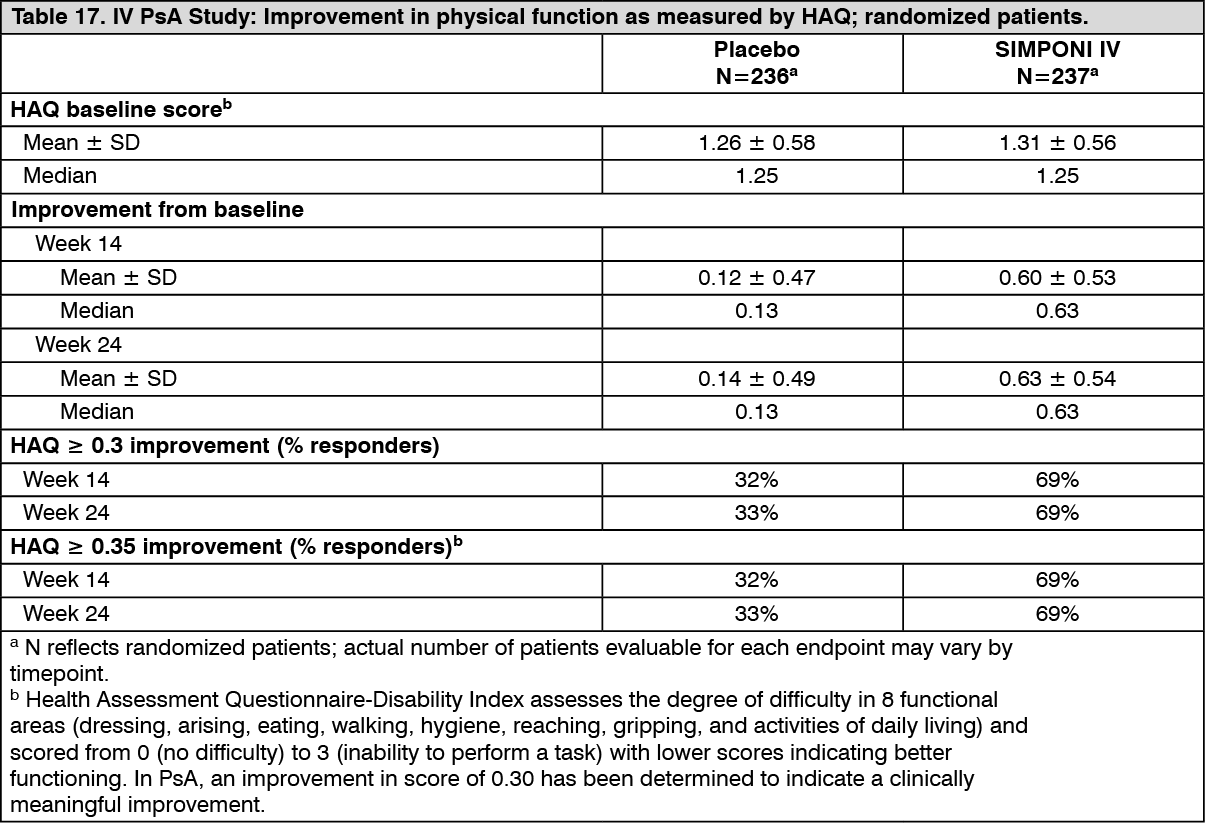 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image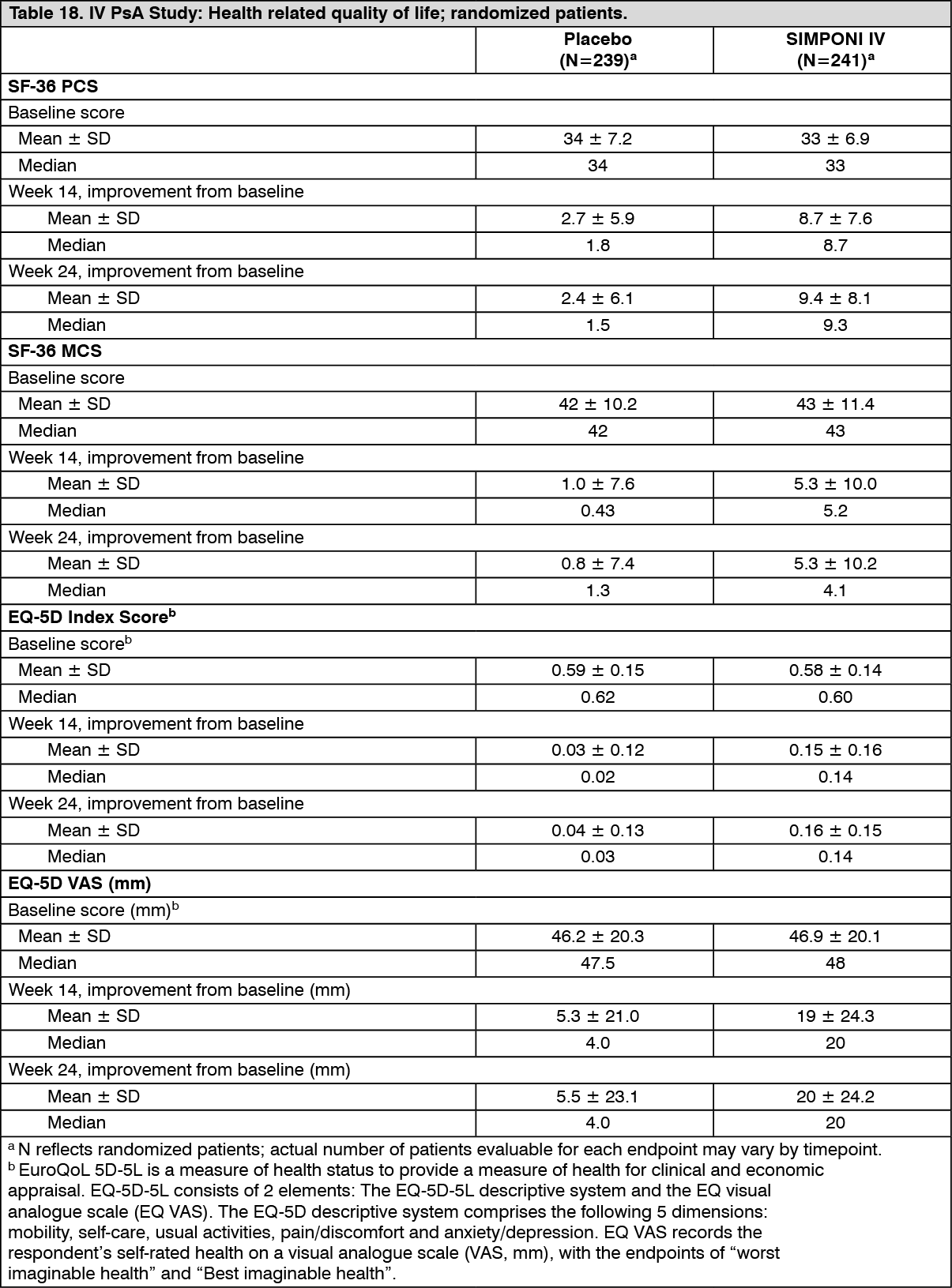 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image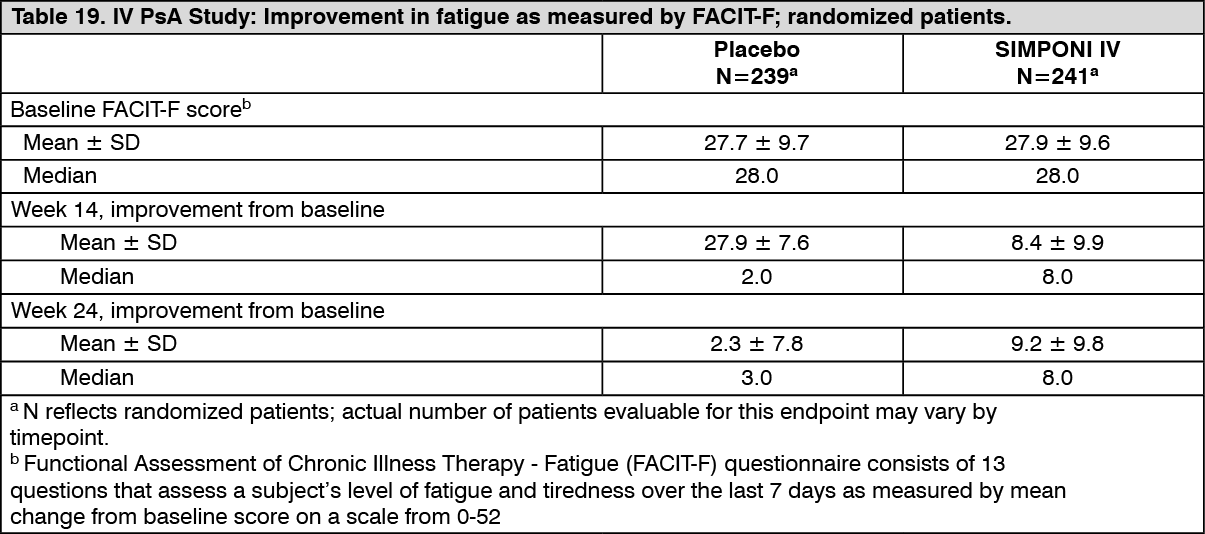 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image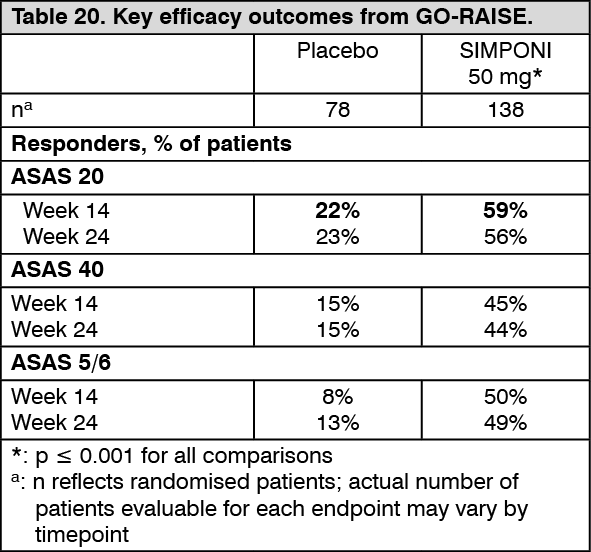 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image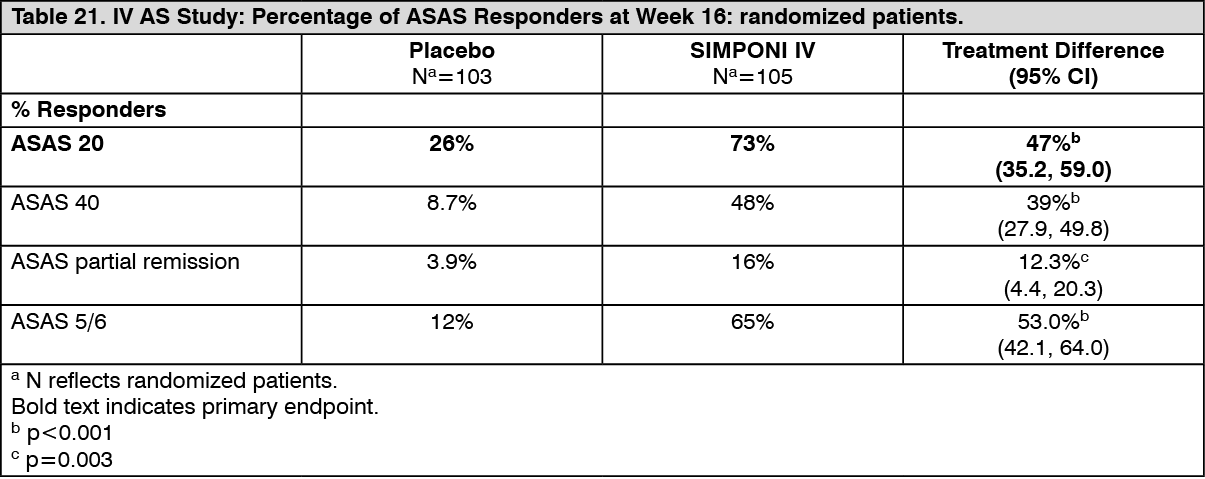 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image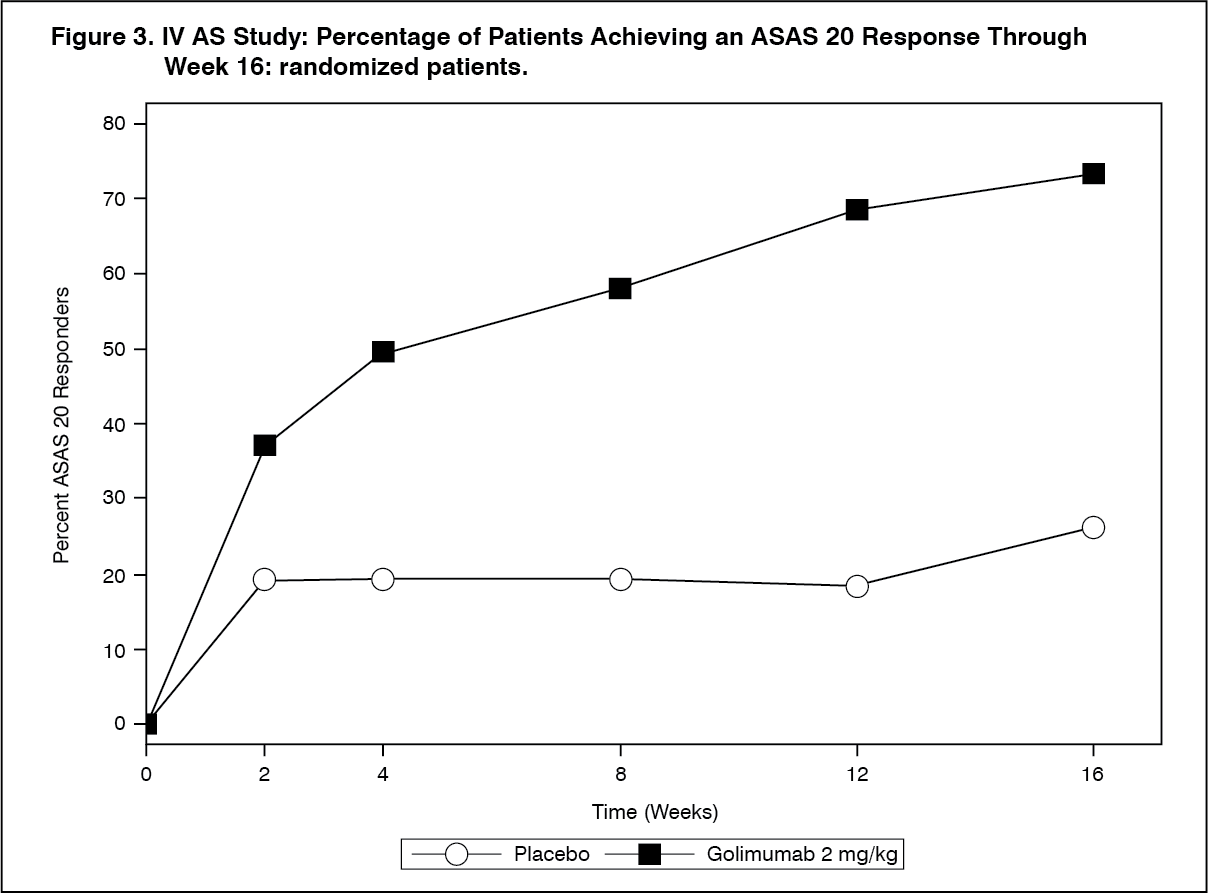 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image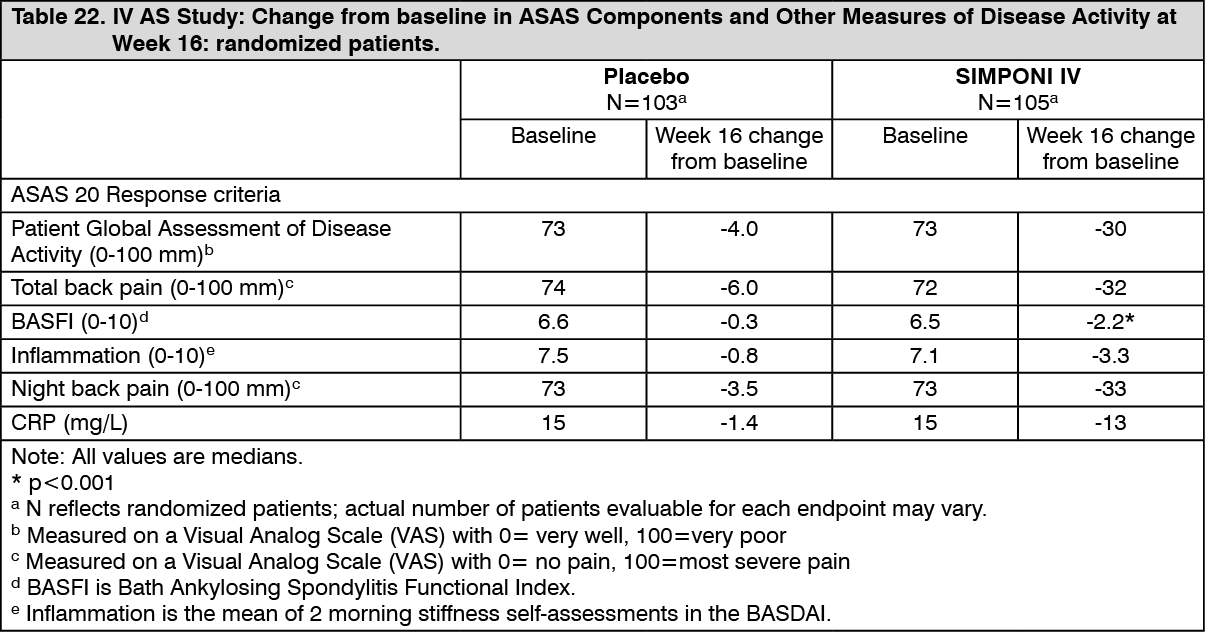 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image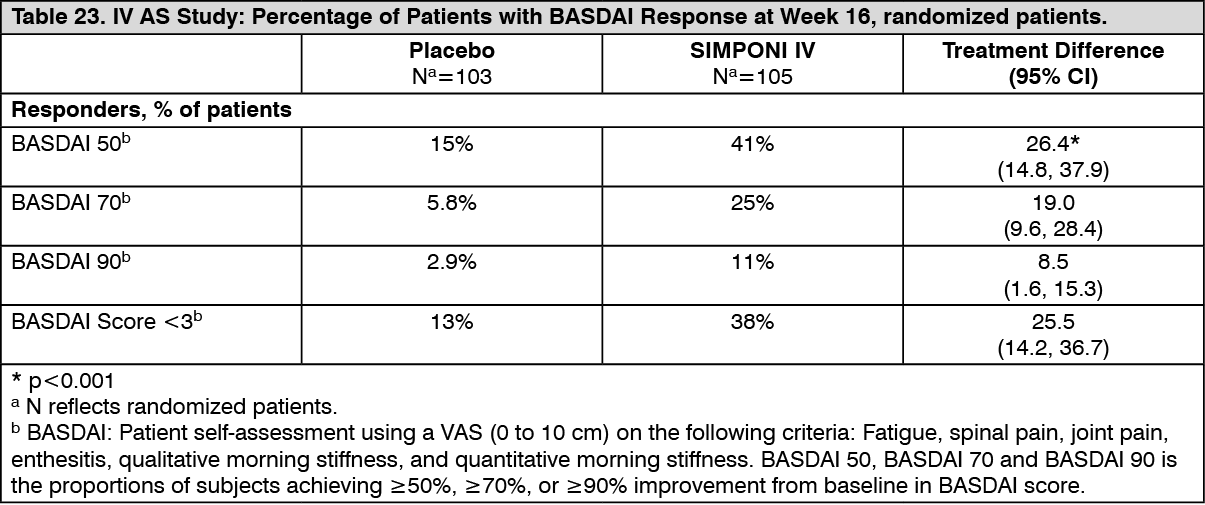 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image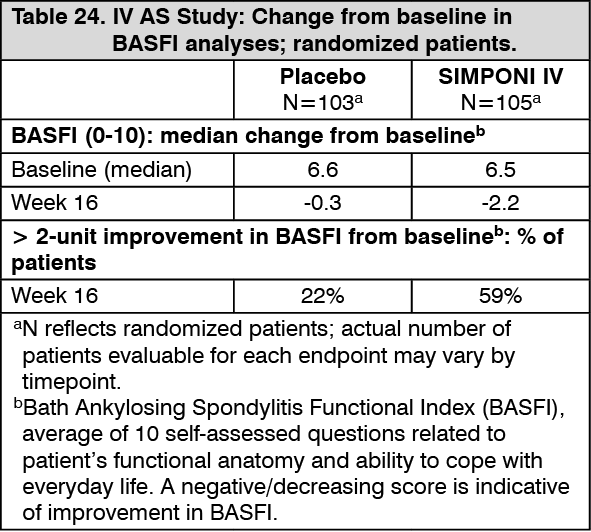 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image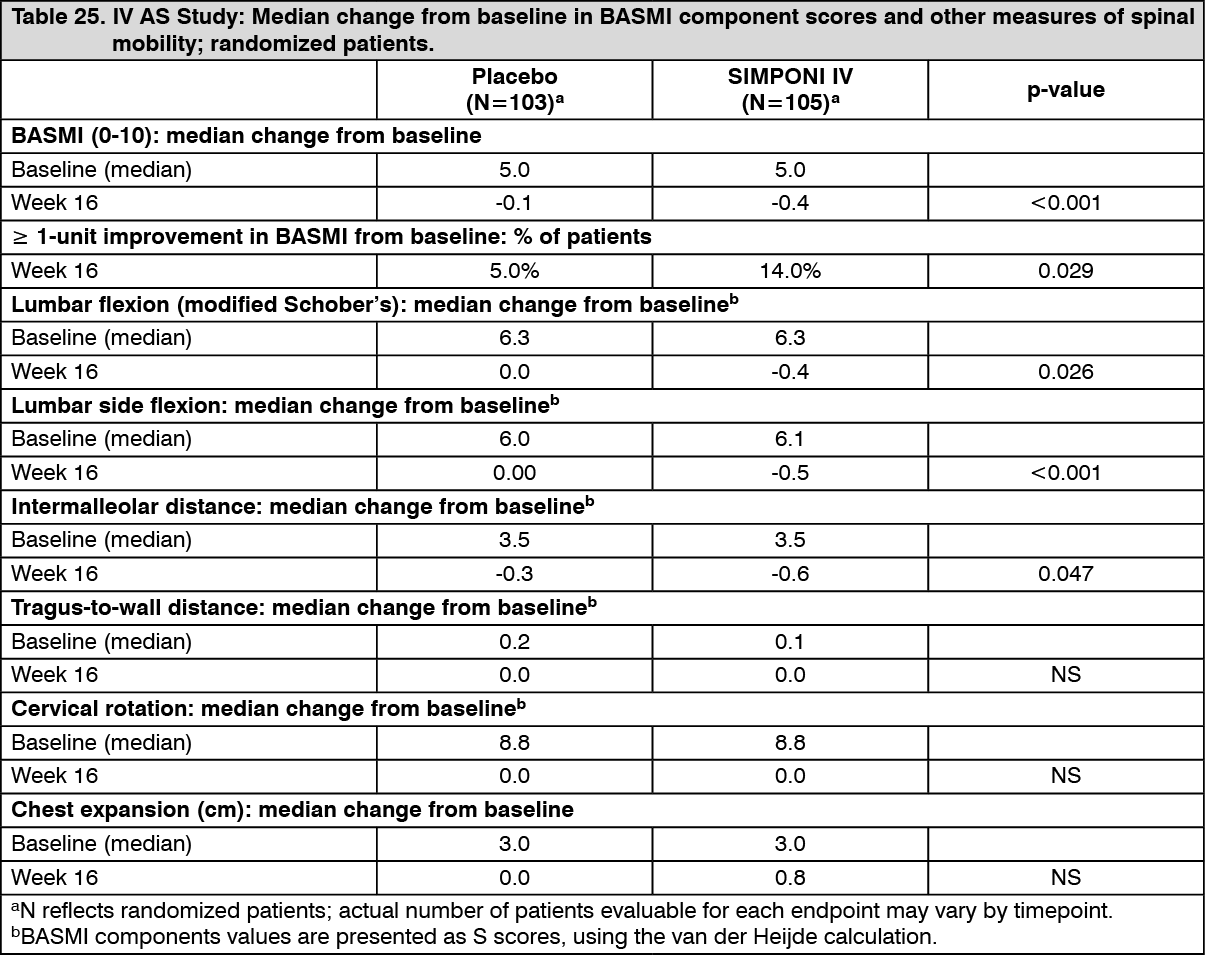 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image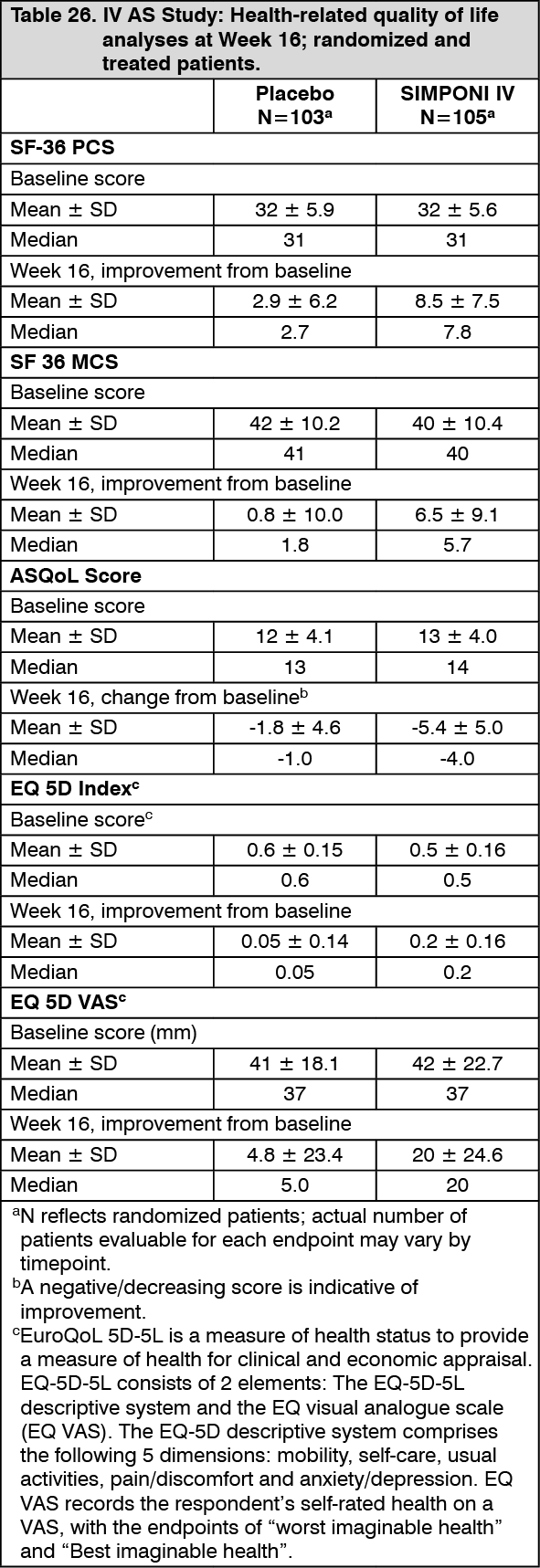 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image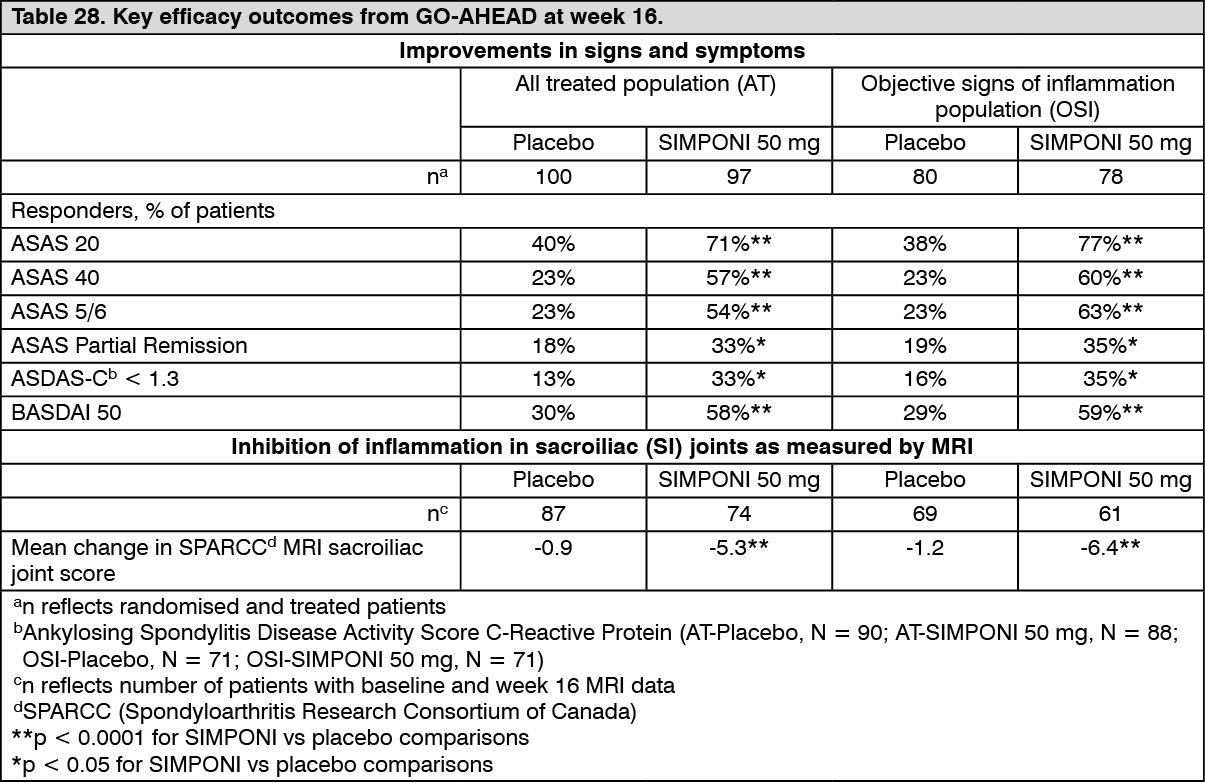 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image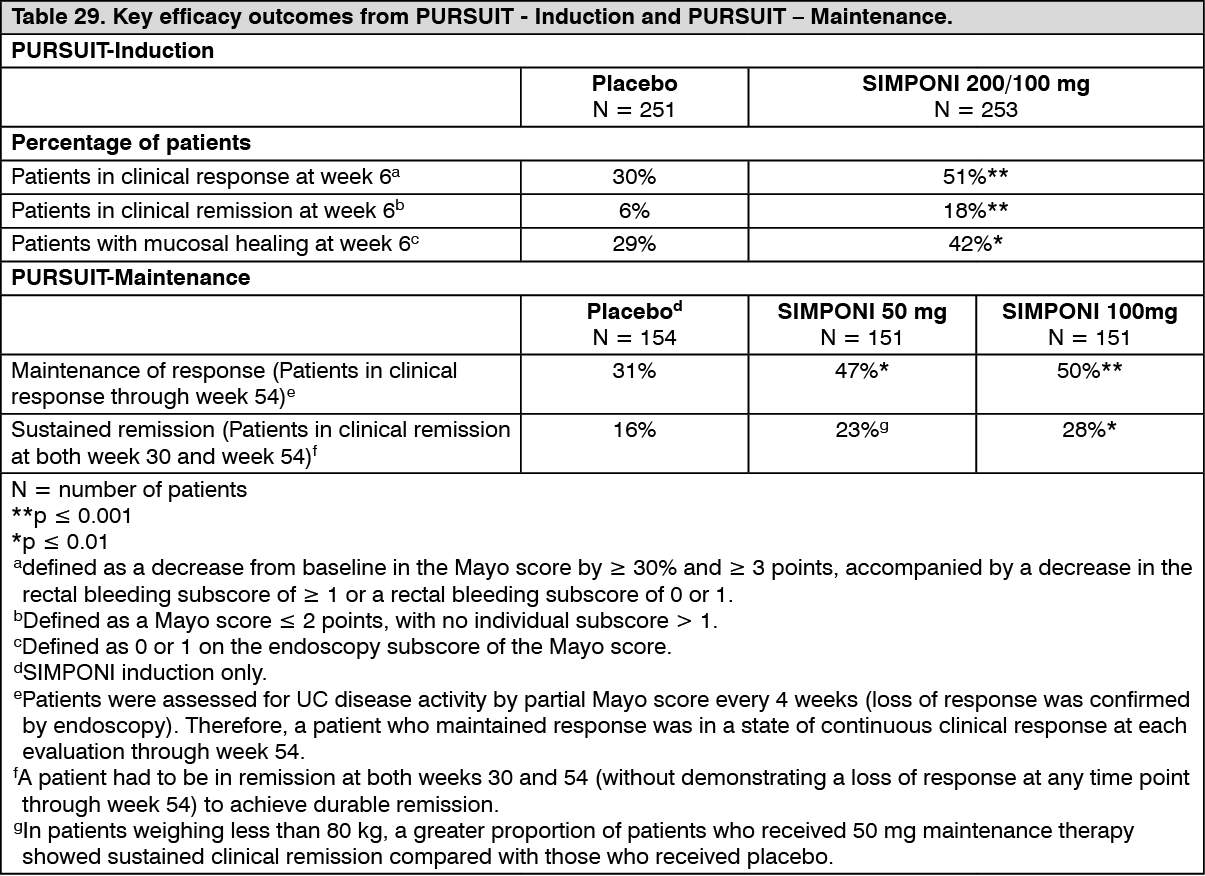 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image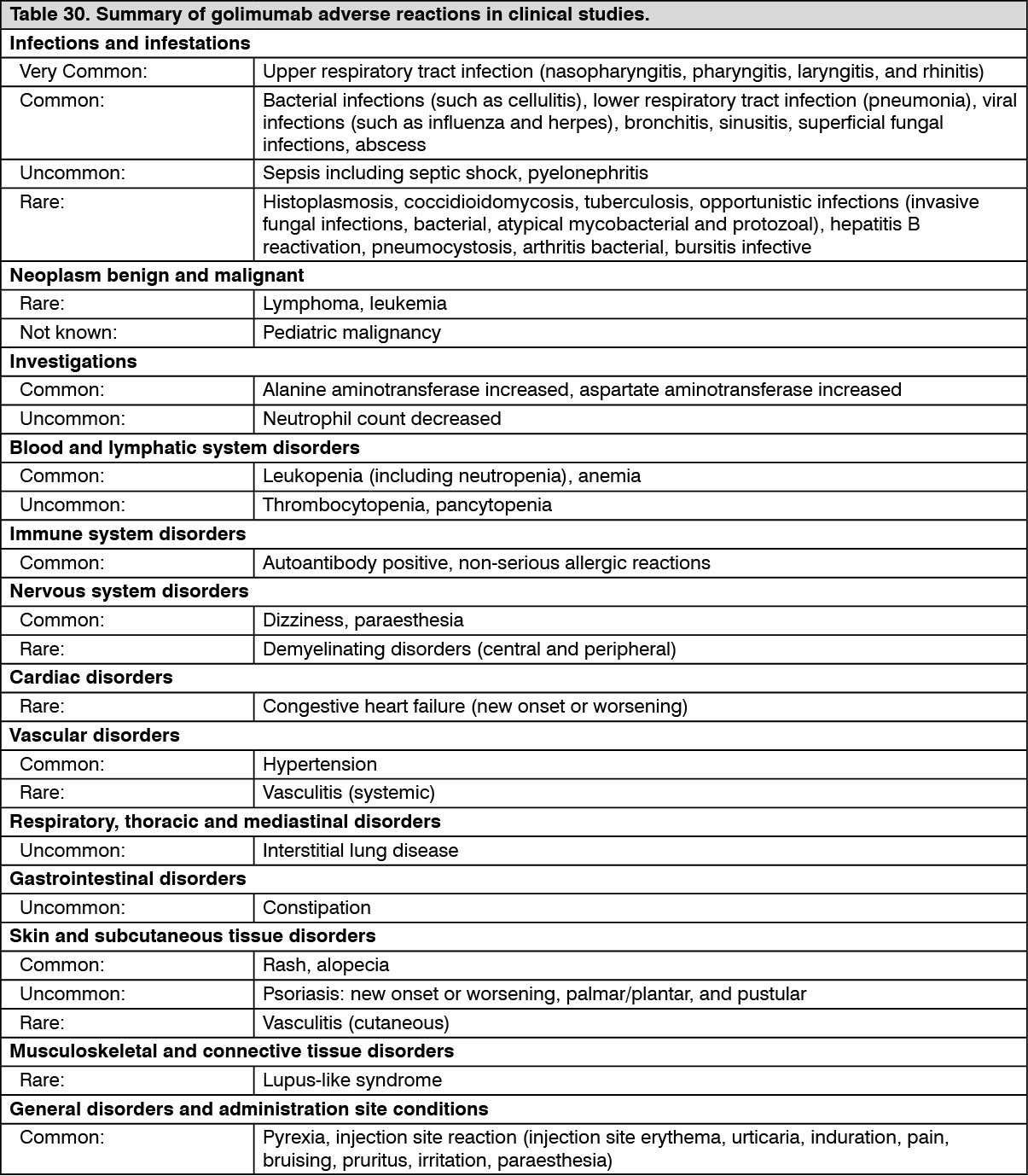 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image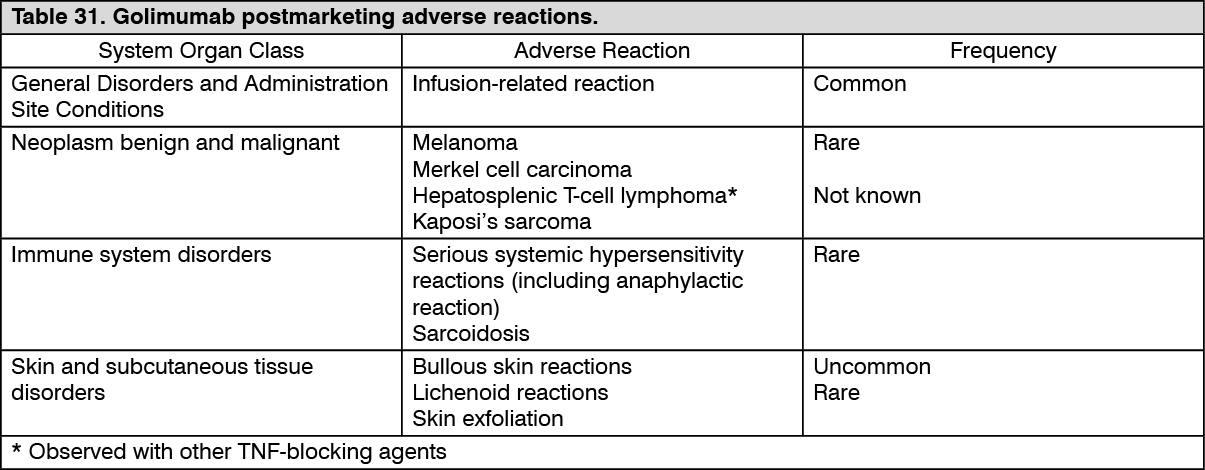 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image 50 mg_0.5 mLdafc195a-6778-4e45-a7a8-a7ac00ae3628.GIF)

 Sign Out
Sign Out
