Pharmacotherapeutic group: EGFR and MET inhibitor.
ATC code: not yet assigned.
Pharmacology: Pharmacodynamics: Mechanism of action: Amivantamab is a low-fucose, fully-human IgG1-based EGFR-MET bispecific antibody with immune cell-directing activity that targets tumors with activating and resistance EGFR mutations and MET mutations and amplifications. Amivantamab binds to the extracellular domains of EGFR and MET.
Preclinical studies show amivantamab is active against tumors with primary EGFR activating mutations such as Exon 19 deletions, L858R substitution, and Exon 20 insertion mutations; secondary EGFR resistance mutations such as T790M and C797S; and resistance to EGFR inhibition due to activation of the MET pathway. Amivantamab disrupts EGFR and MET signaling functions through blocking ligand binding and enhancing degradation of EGFR and MET, thereby preventing tumor growth and progression. The presence of EGFR and MET on the surface of tumor cells also allows for targeting of these cells for destruction by immune effector cells, such as natural killer cells and macrophages, through antibody-dependent cellular cytotoxicity (ADCC) and trogocytosis mechanisms, respectively.
Pharmacodynamic effects: Albumin: Amivantamab decreased serum albumin concentration, a pharmacodynamic effect of MET inhibition, typically during the first 8 weeks; thereafter, albumin concentration stabilized for the remainder of amivantamab treatment.
Immunogenicity: As with all therapeutic proteins, there is the potential for immunogenicity. In a clinical trial of patients with locally advanced or metastatic NSCLC treated with RYBREVANT, 3 (1%) of the 286 evaluable patients tested positive for anti-amivantamab antibodies.
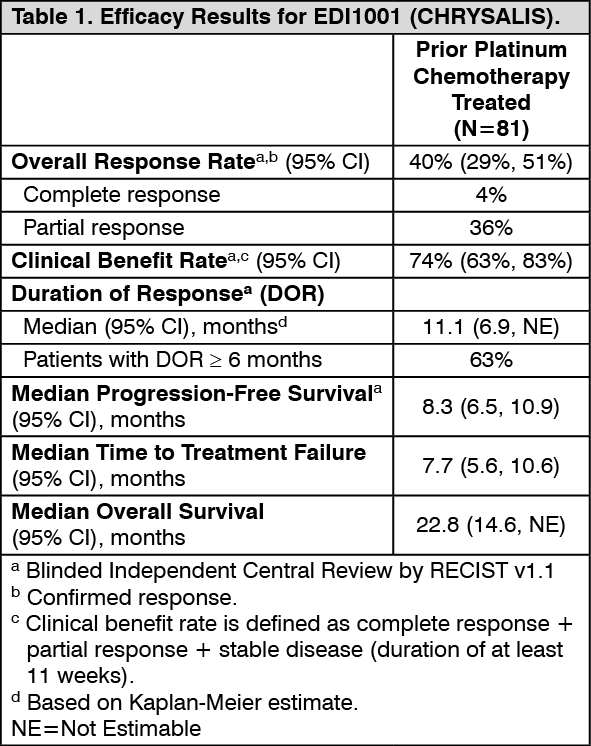
Clinical studies: Locally advanced or metastatic NSCLC with exon 20 insertion mutations: EDI1001 (CHRYSALIS) is a multicenter, open-label, multi-cohort study conducted to assess the safety and efficacy of RYBREVANT in subjects with locally advanced or metastatic NSCLC. Efficacy was evaluated in 81 subjects with locally advanced or metastatic NSCLC who had EGFR Exon 20 insertion mutations as determined by previous local standard of care testing, whose disease had progressed on or after platinum-based chemotherapy, and who had median follow-up of 9.7 months. RYBREVANT was administered intravenously at 1050 mg for subjects <80 kg or 1400 mg for subjects ≥80 kg once weekly for 4 weeks, then every 2 weeks starting at Week 5 until disease progression or unacceptable toxicity.
The median age was 62 (range: 42-84) years, with 9% of the subjects ≥75 years of age; 59% were female; and 49% were Asian and 37% were White. The median number of prior therapies was 2 (range: 1 to 7 therapies). At baseline, 99% had Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 (99%); 53% never smoked; 75% had Stage IV cancer; and 22% had previous treatment for brain metastases. Insertions in Exon 20 were observed at 8 different residues; the most common residues were A767 (24%), S768 (16%), D770 (11%), and N771 (11%).
Efficacy results are summarized in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Anti-tumor activity was observed across all mutation variants.
Pharmacokinetics: Amivantamab area under the concentration-time curve (AUC
1week) increases proportionally over a dose range from 350 to 1750 mg (0.33 to 1.67 times the recommended dose for subjects <80 kg and 0.25 to 1.25 times the recommended dose for subjects ≥80 kg).
Following administration of RYBREVANT at the recommended dose and schedule, the mean ± SD serum maximal concentration (C
max) was 836 ± 264 mcg/mL at 1050 mg for subjects <80 kg and 655 ± 109 mcg/mL at 1400 mg for subjects ≥80 kg at the end of weekly dosing following the fifth dose. The mean ± SD AUC
1week following the fifth dose was 94,946 ± 35,440 mcg.h/mL at 1050 mg for subjects <80 kg and 76,946 ± 14,557 mcg.h/mL at 1400 mg for subjects ≥80 kg. The mean serum AUC
1week was approximately 2.9-fold higher after the fifth dose following the weekly dosing compared to the first dose.
When RYBREVANT was administered, amivantamab steady state was achieved approximately 2 months into the every 2-week dosing period (by the ninth infusion) at 1050 mg, and amivantamab mean ± SD ratio of AUC
1week at steady state to AUC
1week after the first dose was 2.44 ± 0.54.
Distribution: Amavintamab mean ± SD volume of distribution estimated from population PK parameters was 5.13 ± 1.78 L following administration of the recommended dose of RYBREVANT.
Elimination: Amivantamab clearance decreased with increasing dose and with multiple dosing. The mean ± SD linear clearance was estimated to be 360 ± 144 mL/day and the mean ± SD estimated terminal half-life associated with linear clearance estimated from population PK parameter estimates was 11.3 ± 4.53 days following administration of the recommended dose of RYBREVANT.
Special populations: Pediatrics (17 years of age and younger): The pharmacokinetics of RYBREVANT in pediatric patients have not been investigated.
Elderly (65 years of age and older): No clinically meaningful differences in the pharmacokinetics of amivantamab were observed based on age (32-87 years).
Renal impairment: No clinically meaningful effect on the pharmacokinetics of amivantamab was observed in patients with mild (60 ≤ creatinine clearance [CrCl] <90 mL/min) and moderate (29 ≤ CrCl <60 mL/min) renal impairment. The effect of severe renal impairment (15 ≤ CrCl <29 mL/min) on amivantamab pharmacokinetics is unknown.
Hepatic impairment: Changes in hepatic function are unlikely to have any effect on the elimination of amivantamab since IgG1-based molecules such as amivantamab are not metabolized through hepatic pathways.
No clinically meaningful effect in the pharmacokinetics of amivantamab was observed based on mild hepatic impairment [(total bilirubin ≤ ULN and AST > ULN) or (ULN < total bilirubin ≤1.5 x ULN)]. The effect of moderate (total bilirubin 1.5 to 3 times ULN) and severe (total bilirubin >3 times ULN) hepatic impairment on amivantamab pharmacokinetics is unknown.
Gender: The clearance of amivantamab was 24% higher in males than in females; however, no clinically meaningful differences in the pharmacokinetics of amivantamab were observed based on gender.
Weight: The central volume of distribution and clearance of amivantamab increased with increasing body weight. Similar amivantamab exposures were achieved at the recommended dose of RYBREVANT in patients with a body weight <80 kg who received 1050 mg and patients with a body weight ≥80 kg who received 1400 mg.
Toxicology: Non-clinical Information: In repeat-dose toxicity studies in cynomolgus monkeys, amivantamab was well-tolerated at weekly doses up to 120 mg/kg intravenously for 6 weeks or 3 months (~6-8x C
max and ~5-7x AUC human exposure for 1050 and 1400 mg intravenous doses). There were no effects on cardiovascular, respiratory, and nervous system function. Clinical pathology demonstrated non-adverse elevations in serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and globulins, and non-adverse decreases in albumin when compared to the control group. All these values returned to normal ranges in recovery groups. A subcutaneous local tolerance study showed that amivantamab was well tolerated at injection sites in cynomolgus monkeys administered two 125 mg/kg weekly doses.
Carcinogenicity and Mutagenicity: No animal studies have been performed to establish the carcinogenic potential of amivantamab. Routine genotoxicity and carcinogenicity studies are generally not applicable to biologic pharmaceuticals as large proteins cannot diffuse into cells and cannot interact with DNA or chromosomal material.
Reproductive Toxicology: No reproductive toxicology studies have been performed to evaluate the potential effects of amivantamab.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image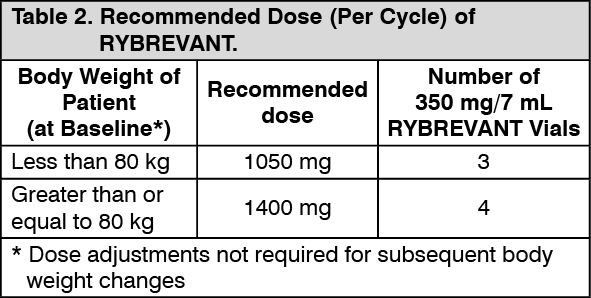 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image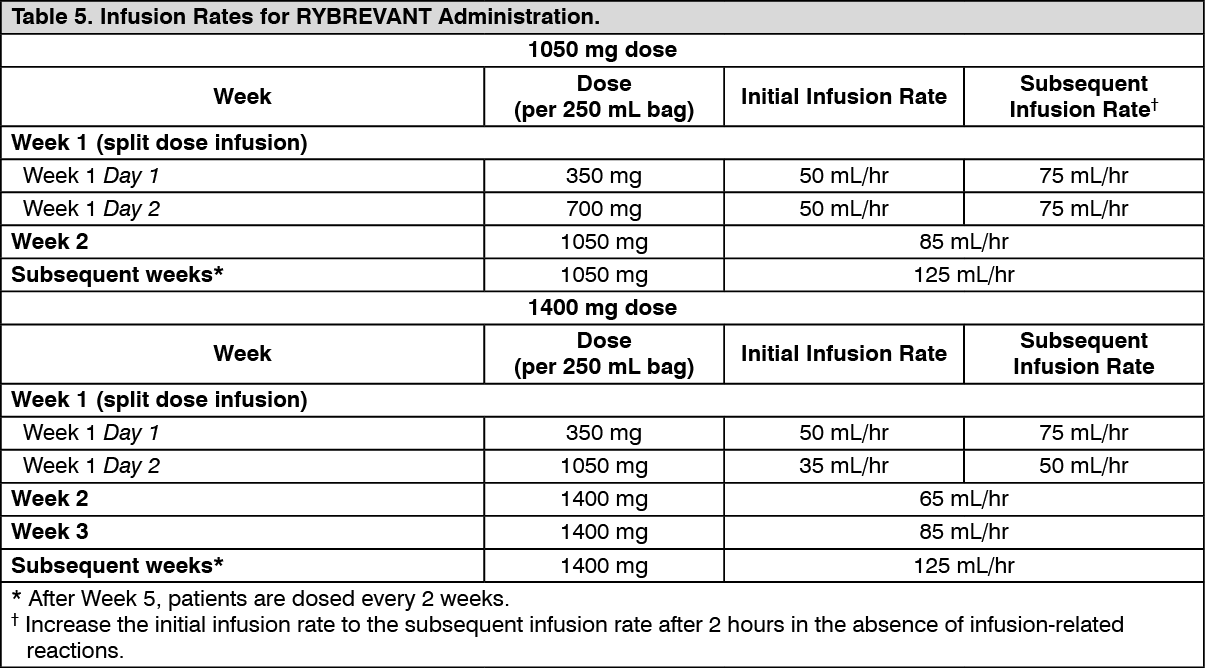 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
