Each Remeron SolTab 15 mg orodispersible tablet contains 15 mg of mirtazapine.
Each Remeron SolTab 30 mg orodispersible tablet contains 30 mg of mirtazapine.
Excipients/Inactive Ingredients: Each Remeron SolTab 15 mg orodispersible tablet contains 4.65 mg aspartame and about 28 mg sucrose.
Each Remeron SolTab 30 mg orodispersible tablet contains 9.30 mg aspartame and about 56 mg sucrose.
Sugar spheres, hypromellose, povidone K30, magnesium stearate, basic butylated methacrylate copolymer, aspartame (E951), citric acid anhydrous, crospovidone (type A), mannitol (E421), microcrystalline cellulose, natural and artificial orange flavor (No. SN027512), sodium bicarbonate.
Pharmacotherapeutic group: Other antidepressants. ATC code: N06AX11.
Pharmacology: Pharmacodynamics: Mirtazapine is a centrally active presynaptic α2-antagonist, which increases central noradrenergic and serotonergic neurotransmission. The enhancement of serotonergic neurotransmission is specifically mediated via 5-HT1 receptors, because 5-HT2 and 5-HT3 receptors are blocked by mirtazapine. Both enantiomers of mirtazapine are presumed to contribute to the antidepressant activity, the S(+) enantiomer by blocking α2 and 5-HT2 receptors and the R(-) enantiomer by blocking 5-HT3 receptors.
The histamine H1-antagonistic activity of mirtazapine is associated with its sedative properties. It has practically no anticholinergic activity and, at therapeutic doses, has only limited effects (e.g. orthostatic hypotension) on the cardiovascular system.
Pharmacokinetics: After oral administration of Remeron, the active substance mirtazapine is rapidly and well absorbed (bioavailability ≈ 50 %), reaching peak plasma levels after approx. two hours. Binding of mirtazapine to plasma proteins is approx. 85 %. The mean half-life of elimination is 20-40 hours; longer half-lives, up to 65 hours, have occasionally been recorded and shorter half-lives have been seen in young men. The half-life of elimination is sufficient to justify once-a-day dosing. Steady state is reached after 3-4 days, after which there is no further accumulation. Mirtazapine displays linear pharmacokinetics within the recommended dose range. Food intake has no influence on the pharmacokinetics of mirtazapine.
Mirtazapine is extensively metabolized and eliminated via the urine and feces within a few days. Major pathways of biotransformation are demethylation and oxidation, followed by conjugation. In vitro data from human liver microsomes indicate that cytochrome P450 enzymes CYP2D6 and CYP1A2 are involved in the formation of the 8-hydroxy metabolite of mirtazapine, whereas CYP3A4 is considered to be responsible for the formation of the N-demethyl and N-oxide metabolites. The demethyl metabolite is pharmacologically active and appears to have the same pharmacokinetic profile as the parent compound.
The clearance of mirtazapine may be decreased as a result of renal or hepatic impairment.
Toxicology: Preclinical safety data: Preclinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, carcinogenicity or genotoxicity.
In reproductive toxicity studies in rats and rabbits no teratogenic effects were observed. At two-fold systemic exposure compared to maximum human therapeutic exposure, there was an increase in post-implantation loss, decrease in the pup birth weights, and reduction in pup survival during the first three days of lactation in rats.
Mirtazapine was not genotoxic in a series of tests for gene mutation and chromosomal and DNA damage. Thyroid gland tumors found in a rat carcinogenicity study and hepatocellular neoplasms found in a mouse carcinogenicity study are considered to be species-specific, non-genotoxic responses associated with long-term treatment with high doses of hepatic enzyme inducers.
Episode of major depression.
In order to prevent crushing the tablet, do not push against the tablet pocket.
Each strip contains six tablet pockets, which are separated by perforations. Tear off one tablet pocket along the dotted lines.
Carefully peel off the lidding foil, starting in the corner indicated by the arrow.
The tablet should be taken out of the strip with dry hands and should be placed on the tongue. The tablet will rapidly disintegrate and can be swallowed without water.
Adults: Treatment should begin with 15 mg daily. The dosage generally needs to be increased to obtain an optimal clinical response. The effective daily dose is usually between 15 and 45 mg. (the dose should be taken at night).
Mirtazapine begins to exert its effect in general after 1-2 weeks of treatment. Treatment with an adequate dose should result in a positive response within 2-4 weeks. With an insufficient response, the dose can be increased up to the maximum dose. If there is no response within a further 2-4 weeks, then treatment should be stopped.
Elderly: The recommended dose is the same as that for adults. In elderly patients an increase in dosing should be done under close supervision to elicit a satisfactory and safe response.
Children and adolescents under the age of 18 years: Remeron should not be used in children and adolescents under the age of 18 years (see Precautions).
Renal impairment: The clearance of mirtazapine may be decreased in patients with moderate to severe renal impairment (creatinine clearance <40 ml/min). This should be taken into account when prescribing Remeron to this category of patients (see Precautions).
Hepatic impairment: The clearance of mirtazapine may be decreased in patients with hepatic impairment. This should be taken into account when prescribing Remeron to this category of patients, particularly with severe hepatic impairment, as patients with severe hepatic impairment have not been investigated (see Precautions).
Mirtazapine has an elimination half-life of 20-40 hours and therefore Remeron is suitable for once daily administration. It should be taken preferably as a single night-time dose before going to bed. Remeron may also be given in two divided doses (once in the morning and once at night-time, the higher dose should be taken at night).
The tablets should be taken orally. The tablet will rapidly disintegrate and can be swallowed without water.
Patients with depression should be treated for a sufficient period of at least 6 months to ensure that the patients are free from symptoms.
It is recommended to discontinue treatment with mirtazapine gradually to avoid withdrawal symptoms (see Precautions).
Present experience concerning overdose with Remeron alone indicates that symptoms are usually mild. Depression of the central nervous system with disorientation and prolonged sedation have been reported, together with tachycardia and mild hyper- or hypotension. However, there is a possibility of more serious outcomes (including fatalities) at dosages much higher than the therapeutic dose, especially with mixed overdoses. In these cases QT prolongation and Torsade de Pointes have also been reported.
Cases of overdose should receive appropriate symptomatic and supportive therapy for vital functions. ECG monitoring should be undertaken. Activated charcoal or gastric lavage should also be considered.
Hypersensitivity to the active substance or to any of the excipients.
Concomitant use of mirtazapine with monoamine oxidase (MAO) inhibitors (see Interactions).
Suicide/suicidal thoughts or clinical worsening: Depression is associated with an increased risk of suicidal thoughts, self harm and suicide (suicide-related events). This risk persists until significant remission occurs. As improvement may not occur during the first few weeks or more of treatment, patients should be closely monitored until such improvement occurs. It is general clinical experience that the risk of suicide may increase in the early stages of recovery.
Patients with a history of suicide-related events or those exhibiting a significant degree of suicidal ideation prior to commencement of treatment are known to be at greater risk of suicidal thoughts or suicide attempts, and should receive careful monitoring during treatment. A meta-analysis of placebo-controlled clinical trials of antidepressants in adult patients with psychiatric disorders showed an increased risk of suicidal behavior with antidepressants compared to placebo in patients less than 25 years old.
Close supervision of patients and in particular those at high risk should accompany therapy with antidepressants especially in early treatment and following dose changes. Patients (and caregivers of patients) should be alerted about the need to monitor for any clinical worsening, suicidal behavior or thoughts and unusual changes in behavior and to seek medical advice immediately if these symptoms present.
With regard to the chance of suicide, in particular at the beginning of treatment, only the smallest amount of Remeron SolTab orodispersible tablets should be given to the patient consistent with good patient management, in order to reduce the risk of overdose.
Bone marrow depression: Bone marrow depression, usually presenting as granulocytopenia or agranulocytosis, has been reported during treatment with Remeron. Reversible agranulocytosis has been reported as a rare occurrence in clinical studies with Remeron. In the postmarketing period with Remeron very rare cases of agranulocytosis have been reported, mostly reversible, but in some cases fatal. Fatal cases mostly concerned patients with an age above 65. The physician should be alert for symptoms like fever, sore throat, stomatitis or other signs of infection; when such symptoms occur, treatment should be stopped and blood counts taken.
Jaundice: Treatment should be discontinued if jaundice occurs.
Conditions which need supervision: Careful dosing as well as regular and close monitoring is necessary in patients with: Epilepsy and organic brain syndrome: Although clinical experience indicates that epileptic seizures are rare during mirtazapine treatment, as with other antidepressants, Remeron should be introduced cautiously in patients who have a history of seizures. Treatment should be discontinued in any patient who develops seizures, or where there is an increase in seizure frequency.
Hepatic impairment: Following a single 15 mg oral dose of mirtazapine, the clearance of mirtazapine was approximately 35 % decreased in mild to moderate hepatically impaired patients, compared to subjects with normal hepatic function. The average plasma concentration of mirtazapine was about 55 % increased.
Renal impairment: Following a single 15 mg oral dose of mirtazapine, in patients with moderate (10 ml/min ≤ creatinine clearance < 40 ml/min) and severe (creatinine clearance < 10 ml/min) renal impairment the clearance of mirtazapine was about 30 % and 50 % decreased respectively, compared to normal subjects. The average plasma concentration of mirtazapine was about 55 % and 115 % increased respectively. No significant differences were found in patients with mild renal impairment (40 ml/min ≤ creatinine clearance < 80 ml/min) as compared to the control group.
Cardiac diseases like conduction disturbances, angina pectoris and recent myocardial infarction, where normal precautions should be taken and concomitant medicines carefully administered.
Low blood pressure.
Diabetes mellitus: In patients with diabetes, antidepressants may alter glycemic control. Insulin and/or oral hypoglycemic dosage may need to be adjusted and close monitoring is recommended.
Like with other antidepressants, the following should be taken into account: Worsening of psychotic symptoms can occur when antidepressants are administered to patients with schizophrenia or other psychotic disturbances; paranoid thoughts can be intensified.
When the depressive phase of bipolar disorder is being treated, it can transform into the manic phase. Patients with a history of mania/hypomania should be closely monitored. Mirtazapine should be discontinued in any patient entering a manic phase.
Although Remeron is not addictive, post-marketing experience shows that abrupt termination of treatment after long term administration may sometimes result in withdrawal symptoms. The majority of withdrawal reactions are mild and self-limiting. Among the various reported withdrawal symptoms, dizziness, agitation, anxiety, headache and nausea are the most frequently reported. Even though they have been reported as withdrawal symptoms, it should be realized that these symptoms may be related to the underlying disease. As advised in Dosage & Administration, it is recommended to discontinue treatment with mirtazapine gradually.
Care should be taken in patients with micturition disturbances like prostate hypertrophy and in patients with acute narrow-angle glaucoma and increased intra-ocular pressure (although there is little chance of problems with Remeron because of its very weak anticholinergic activity).
Akathisia/psychomotor restlessness: The use of antidepressants have been associated with the development of akathisia, characterized by a subjectively unpleasant or distressing restlessness and need to move often accompanied by an inability to sit or stand still. This is most likely to occur within the first few weeks of treatment. In patients who develop these symptoms, increasing the dose may be detrimental.
The effect of Remeron on QTCc interval was assessed in a randomized, placebo and moxifloxacin controlled clinical trial involving 54 healthy volunteers using exposure response analysis. This trial revealed that both 45mg (therapeutic) and 75mg (supratherapeutic) doses of mirtazapine did not affect the QTCc interval to a clinically meaningful extent. During the post-marketing use of mirtazapine, cases of QT prolongation, Torsades de Pointes, ventricular tachycardia, and sudden death, have been reported. The majority of reports occurred in association with overdose or in patients with other risk factors for QT prolongation, including concomitant use of QTc prolonging medicines (see Overdosage and Interactions). Caution should be exercised when Remeron is prescribed in patients with known cardiovascular disease or family history of QT prolongation, and in concomitant use with other medicinal products thought to prolong the QTc interval.
Hyponatremia: Hyponatremia has been reported very rarely with the use of mirtazapine. Caution should be exercised in patients at risk, such as elderly patients or patients concomitantly treated with medications known to cause hyponatremia.
Serotonin syndrome: Interaction with serotonergic active substances: serotonin syndrome may occur when selective serotonin reuptake inhibitors (SSRIs) are used concomitantly with other serotonergic active substances (see Interactions). Symptoms of serotonin syndrome may be hyperthermia, rigidity, myoclonus, autonomic instability with possible rapid fluctuations of vital signs, mental status changes that include confusion, irritability and extreme agitation progressing to delirium and coma. Caution should be advised and a closer clinical monitoring is required when these active substances are combined with mirtazapine. Treatment with Remeron should be discontinued if such events occur and supportive symptomatic treatment initiated. From post marketing experience it appears that serotonin syndrome occurs very rarely in patients treated with Remeron alone (see Adverse Reactions).
Sucrose: Remeron SolTab contains sugar spheres, containing sucrose. Patients with rare hereditary problems of fructose intolerance, glucose-galactose malabsorption or sucrase-isomaltase insufficiency should not take this medicine.
Aspartame: Remeron SolTab contains aspartame, a source of phenylalanine. Each tablet with 15 mg, 30 mg and 45 mg mirtazapine corresponds to 2.6 mg, 5.2 mg and 7.8 mg phenylalanine, respectively. It may be harmful for patients with phenylketonuria.
Effects on ability to drive and use machines: Remeron has minor or moderate influence on the ability to drive and use machines. Remeron may impair concentration and alertness (particularly in the initial phase of treatment). Patients should avoid the performance of potentially dangerous tasks, which require alertness and good concentration, such as driving a motor vehicle or operating machinery, at any time when affected.
Use in children and adolescents under 18 years of age: Remeron should not be used in the treatment of children and adolescents under the age of 18 years. Suicide-related behaviors (suicide attempt and suicidal thoughts), and hostility (predominantly aggression, oppositional behavior and anger) were more frequently observed in clinical trials among children and adolescents treated with antidepressants compared to those treated with placebo. If, based on clinical need, a decision to treat is nevertheless taken, the patient should be carefully monitored for the appearance of suicidal symptoms. In addition, long-term safety data in children and adolescents concerning growth, maturation and cognitive and behavioral development are lacking.
Use in elderly: Elderly patients are often more sensitive, especially with regard to the undesirable effects of antidepressants. During clinical research with Remeron, undesirable effects have not been reported more often in elderly patients than in other age groups.
Limited data of the use of mirtazapine in pregnant women do not indicate an increased risk for congenital malformations. Studies in animals have not shown any teratogenic effects of clinical relevance, however developmental toxicity has been observed (see Pharmacology: Toxicology: Preclinical Safety Data under Actions). Caution should be exercised when prescribing to pregnant women. If Remeron is used until, or shortly before birth, postnatal monitoring of the newborn is recommended to account for possible discontinuation effects.
Animal studies and limited human data have shown excretion of mirtazapine in breast milk only in very small amounts. A decision on whether to continue/discontinue breast-feeding or to continue/discontinue therapy with Remeron should be made taking into account the benefit of breast-feeding to the child and the benefit of Remeron therapy to the woman.
Depressed patients display a number of symptoms that are associated with the illness itself. It is therefore sometimes difficult to ascertain which symptoms are a result of the illness itself and which are a result of treatment with Remeron.
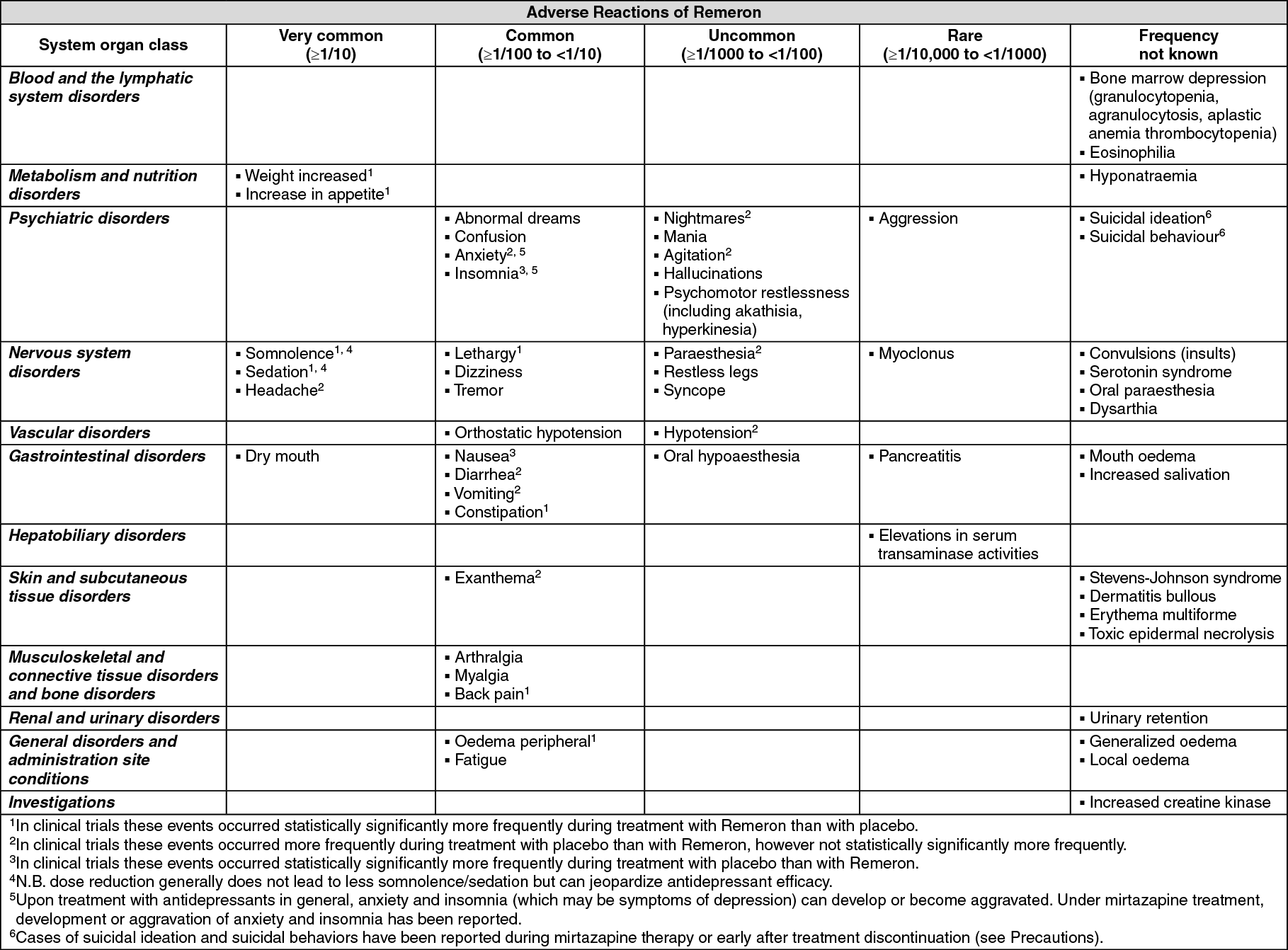
The most commonly reported adverse reactions, occurring in more than 5 % of patients treated with Remeron in randomized placebo-controlled trials (see table) are somnolence, sedation, dry mouth, weight increased, increase in appetite, dizziness and fatigue.
All randomized placebo-controlled trials in patients (including indications other than major depressive disorder), have been evaluated for adverse reactions of Remeron. The meta-analysis considered 20 trials, with a planned duration of treatment up to 12 weeks, with 1501 patients (134 person years) receiving doses of mirtazapine up to 60 mg and 850 patients (79 person years) receiving placebo. Extension phases of these trials have been excluded to maintain comparability to placebo treatment.
The table shows the categorized incidence of the adverse reactions, which occurred in the clinical trials statistically significantly more frequently during treatment with Remeron than with placebo, added with adverse reactions from spontaneous reporting. The frequencies of the adverse reactions from spontaneous reporting are based on the reporting rate of these events in the clinical trials. The frequency of adverse reactions from spontaneous reporting for which no cases in the randomized placebo-controlled patient trials were observed with mirtazapine has been classified as 'not known'.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In laboratory evaluations in clinical trials transient increases in transaminases and gamma-glutamyl transferase have been observed (however associated adverse events have not been reported statistically significantly more frequently with Remeron than with placebo).
Pharmacodynamic interactions: Mirtazapine should not be administered concomitantly with MAO inhibitors or within two weeks after discontinuation of MAO inhibitor therapy. In the opposite way about two weeks should pass before patients treated with mirtazapine should be treated with MAO inhibitors (see Contraindications). In addition, as with SSRIs, co-administration with other serotonergic active substances (L-tryptophan, triptans, tramadol, linezolid, methylene blue, SSRIs, venlafaxine, lithium and St. John's Wort – Hypericum perforatum – preparations) may lead to an incidence of serotonin associated effects (see Serotonin Syndrome under Precautions).
Mirtazapine may increase the sedating properties of benzodiazepines and other sedatives (notably most antipsychotics, histamine H1 antagonists, opioids). Caution should be exercised when these medicinal products are prescribed together with mirtazapine.
Mirtazapine may increase the CNS depressant effect of alcohol. Patients should therefore be advised to avoid alcoholic beverages while taking mirtazapine.
Mirtazapine dosed at 30 mg once daily caused a small but statistically significant increase in the international normalized ratio (INR) in subjects treated with warfarin. As at a higher dose of mirtazapine a more pronounced effect cannot be excluded, it is advisable to monitor the INR in case of concomitant treatment of warfarin with mirtazapine.
The risk of QT prolongation and/or ventricular arrhythmias (e.g. Torsades de Pointes) may be increased with concomitant use of medicines which prolong the QTc interval (e.g. some antipsychotics and antibiotics) and in case of mirtazapine overdose.
Pharmacokinetic interactions: Carbamazepine and phenytoin, CYP3A4 inducers, increased mirtazapine clearance about twofold, resulting in a decrease in average plasma mirtazapine concentration of 60 % and 45 %, respectively. When carbamazepine or any other inducer of hepatic metabolism (such as rifampicin) is added to mirtazapine therapy, the mirtazapine dose may have to be increased. If treatment with such medicinal product is discontinued, it may be necessary to reduce the mirtazapine dose.
Co-administration of the potent CYP3A4 inhibitor ketoconazole increased the peak plasma levels and the AUC of mirtazapine by approximately 40% and 50% respectively.
When cimetidine (weak inhibitor of CYP1A2, CYP2D6 and CYP3A4) is administered with mirtazapine, the mean plasma concentration of mirtazapine may increase more than 50 %. Caution should be exercised and the dose may have to be decreased when co-administering mirtazapine with potent CYP3A4 inhibitors, HIV protease inhibitors, azole antifungals, erythromycin, cimetidine or nefazodone.
Interaction studies did not indicate any relevant pharmacokinetic effects on concurrent treatment of mirtazapine with paroxetine, amitriptyline, risperidone or lithium.
Special precautions for disposal and other handling: No special requirements.
Incompatibilities: Not applicable.
Store in the original package in order to protect from light and moisture.
Shelf-Life: Refer to outer carton.
N06AX11 - mirtazapine ; Belongs to the class of other antidepressants.
Remeron SolTab 15 mg
30's
Remeron SolTab 30 mg
30's


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
