


 Sign Out
Sign Out


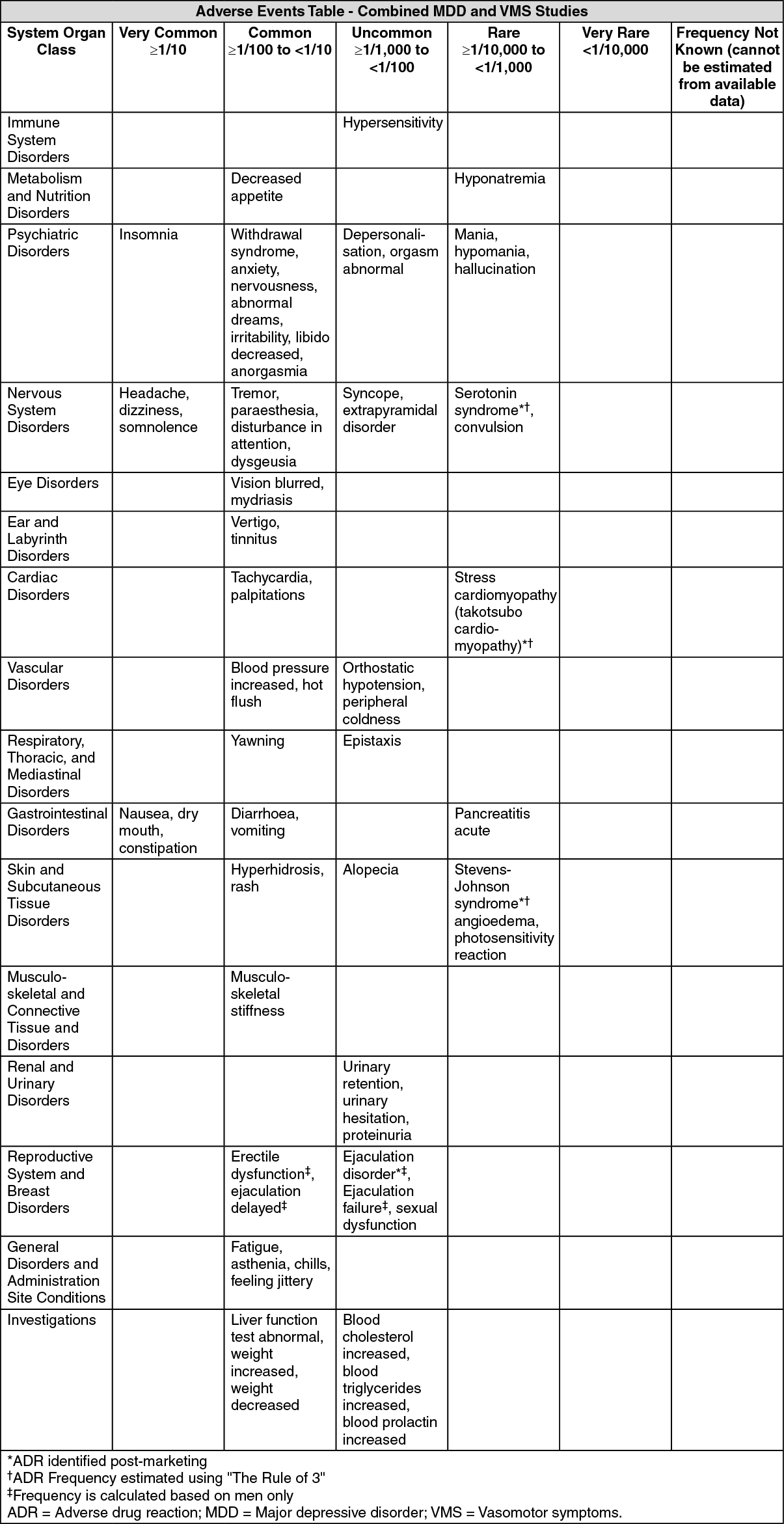
ADRs by SOC and CIOMS frequency category listed in order of decreasing medical seriousness within each frequency category and SOC. (See table.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIschemic cardiac adverse events: In clinical trials, there were uncommon reports of ischemic cardiac adverse events, including myocardial ischemia, myocardial infarction, and coronary occlusion requiring revascularization; these patients had multiple underlying cardiac risk factors. More patients experienced these events during desvenlafaxine treatment as compared to placebo (see PRECAUTIONS).
Discontinuation symptoms: Adverse drug reactions reported in association with abrupt discontinuation, dose reduction or tapering of treatment in MDD clinical trials at a rate of ≥2% include: dizziness, withdrawal syndrome, nausea and headache. In general, discontinuation symptoms occurred more frequently with higher doses and with longer duration of therapy (see DOSAGE & ADMINISTRATION and PRECAUTIONS).
Adverse reactions leading to discontinuation of therapy: The most common adverse reactions leading to discontinuation in at least 2% of the desvenlafaxine-treated patients in the short-term trials, up to 12 weeks, was: nausea (2%); in the long-term studies, up to 11 months, no events lead to discontinuation in at least 2% of the patients and at a rate greater than placebo in the double-blind phase.
Pediatric patients: In general, the adverse reaction profile of PRISTIQ (in placebo-controlled clinical studies) in children and adolescents (aged 7 to 17) was similar to that seen for adults.
The most frequently reported events from the placebo-controlled studies were: Headache (17.3%), Nausea (8.6%), Abdominal pain upper (8.5%), Nasopharyngitis (5.3%), Dizziness (4.6%), Upper respiratory tract infection (4.2%), Decreased appetite (4.2%), Vomiting (3.9%), Fatigue (3.2%), and Insomnia (3.2%). Of these, events with an incidence in the PRISTIQ groups >2 times that of the placebo group were: Fatigue (4.2% vs. 1.7%), and Insomnia (4.2% vs. 1.7%).
When compared to adverse event rates in adults, the following events occurred more frequently in pediatric patients (incidence of ≥3% in pediatric patients and <3% in adults): Abdominal pain upper, Weight increased, Gastroenteritis viral, Dysmenorrhoea, Accidental overdose, Cough, Irritability, Oropharyngeal pain, and Sinusitis.
As with adults, increased blood pressure, abnormal bleeding, mania/hypomania, discontinuation syndrome, seizure, suicide attempt, suicidal behavior, self-injurious behavior and suicidal ideation were observed in pediatric clinical studies (see Precautions).
Geriatric use: Of the 7,785 patients in MDD clinical trials treated with desvenlafaxine, 5% of patients were 65 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients; however, in the short-term placebo-controlled trials, there was a higher incidence of systolic orthostatic hypotension and in both short-term and long-term placebo-controlled trials, there were increases in systolic blood pressure in patients ≥65 years of age compared to patients <65 years of age treated with desvenlafaxine.
Adverse reactions reported with other SNRIs: Although gastrointestinal bleeding is not considered an adverse reaction for desvenlafaxine succinate, it is an adverse reaction for other SNRIs and may also occur with desvenlafaxine succinate.
View ADR Monitoring Form