Pharmacodynamic group: Antivirals for systemic use.
ATC code: J05AE10.
Pharmacology: Pharmacodynamics: Mechanism of action: Darunavir is an inhibitor of the dimerization and of the catalytic activity of the HIV-1 protease. It selectively inhibits the cleavage of HIV encoded Gag-Pol polyproteins in virus infected cells, thereby preventing the formation of mature infectious virus particles.
Darunavir tightly binds to the HIV-1 protease with a K
D of 4.5 x 10
-12 M. Darunavir shows resilience to the effects of HIV protease inhibitors Resistance-Associated Mutations (RAMs).
Darunavir is not an inhibitor of any of 13 tested human cellular proteases.
Pharmacodynamic effects: Microbiology: Antiviral activity
in vitro: Darunavir exhibits activity against laboratory strains and clinical isolates of HIV-1 and laboratory strains of HIV-2 in acutely infected T-cell lines, human peripheral blood mononuclear cells and human monocytes/macrophages with median EC
50 values ranging from 1.2 to 8.5 nM (0.7 to 5.0 ng/mL). Darunavir demonstrates antiviral activity
in vitro against a broad panel of HIV-1 group M (A, B, C, D, E, F, G) and group O primary isolates with EC
50 values ranging from < 0.1 to 4.3 nM. These EC
50 values are well below the 50% cellular toxicity concentration range of 87 μM to > 100 μM.
The EC
50 value of darunavir increases by a median factor of 5.4 in the presence of human serum. Darunavir showed synergistic antiviral activity when studied in combination with the protease inhibitors ritonavir, nelfinavir, or amprenavir and additive antiviral activity when studied in combination with the protease inhibitors indinavir, saquinavir, lopinavir, atazanavir, or tipranavir, the N(t)RTIs zidovudine, lamivudine, zalcitabine, didanosine, stavudine, abacavir, emtricitabine, or tenofovir, the NNRTIs etravirine, nevirapine, delavirdine, rilpivirine, or efavirenz and the fusion inhibitor enfuvirtide. No antagonism was observed between darunavir and any of those antiretrovirals.
Resistance
in vitro:
In vitro selection of darunavir-resistant virus from wildtype HIV-1 was lengthy (> 3 years). The selected viruses were unable to grow in the presence of darunavir concentrations above 400 nM. Viruses selected in these conditions and showing decreased susceptibility to darunavir (range: 23-50-fold) harbored 2 to 4 amino acid substitutions in the protease gene. The decreased susceptibility to darunavir of the emerging viruses in the selection experiment could not be explained by the emergence of these protease mutations.
In vitro selection of darunavir-resistant HIV-1 (range: 53-641-fold change in EC50 values [FC]) from 9 HIV-1 strains harboring multiple PI RAMs resulted in the overall emergence of 22 mutations in the protease, of which L10F, V32I, L33F, S37N, M46I, I47V, I50V, L63P, A71V, and I84V were present in more than 50% of the 9 darunavir-resistant isolates. A minimum of 8 of these darunavir
in vitro selected mutations, from which at least 2 were already present in the protease prior to selection, were required in the HIV-1 protease to render a virus resistant (FC > 10) to darunavir.
In 1113 clinical isolates resistant to amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and/or tipranavir and in 886 baseline isolates from the patients enrolled in the POWER 1 and POWER 2 trials and in the POWER 3 analysis, only the subgroups with > 10 PI RAMs showed a median FC for darunavir > 10.
Cross-resistance
in vitro: Cross-resistance has been observed among HIV protease inhibitors. Darunavir has a < 10-fold decreased susceptibility against 90% of 3309 clinical isolates resistant to amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and/or tipranavir showing that viruses resistant to most PIs remain susceptible to darunavir.
Seven of the 9 darunavir-resistant viruses selected from PI-resistant viruses had phenotypic data for tipranavir. Six of those showed a FC < 3 for tipranavir, indicative of limited cross-resistance between these 2 protease inhibitors.
Cross-resistance between darunavir and the nucleoside/nucleotide reverse transcriptase inhibitors, the non-nucleoside reverse transcriptase inhibitors, the entry inhibitors, or the integrase inhibitors, is unlikely because the viral targets for those inhibitors are different.
Clinical studies: All clinical trials described in this section were performed with PREZISTA co-administered with low dose ritonavir.
Description of clinical studies in adults: Efficacy of PREZISTA/rtv 600/100 mg b.i.d. in treatment-experienced adult patients: The evidence of efficacy of PREZISTA/rtv 600/100 mg b.i.d. in treatment-experienced patients is based on the analyses of 96 week analysis of the Phase III trial TITAN in treatment-experienced , lopinavir/rtv naive patients and on the analyses of 96 week data from the Phase IIb trials POWER 1, 2 and 3, in patients with high level of PI resistance.
TITAN is a randomised, controlled, open-label Phase III trial comparing PREZISTA/rtv 600/100 mg b.i.d. versus lopinavir/rtv 400/100 mg b.i.d. in antiretroviral treatment-experienced, lopinavir/rtv naive HIV-1 infected adult patients. Both arms used an optimised background regimen (OBR) consisting of at least 2 antiretrovirals (NRTIs with or without NNRTIs). HIV-1 infected patients who were eligible for this trial had plasma HIV-1 RNA >1000 copies/ml and were on a highly active antiretroviral therapy regimen (HAART) for at least 12 weeks.
Virologic response was defined as a confirmed plasma HIV-1 RNA viral load < 400 copies/mL. Analyses included 595 patients in the TITAN trial who had completed 96 weeks of treatment or discontinued earlier.
Demographics and baseline characteristics were balanced between the PREZISTA/rtv arm and the lopinavir/ritonavir arm. The 298 patients on PREZISTA/rtv 600/100 mg b.i.d. had a median age of 40 years (range 18-68), 77% were male, 54% white, 18% black, 15% hispanic, and 9% asian. The mean baseline plasma HIV-1 RNA was 4.33 log10 copies/mL and the median baseline CD4+ cell count was 235 x 10
6 cells/l (range 3 - 831 x 10
6 cells/l).
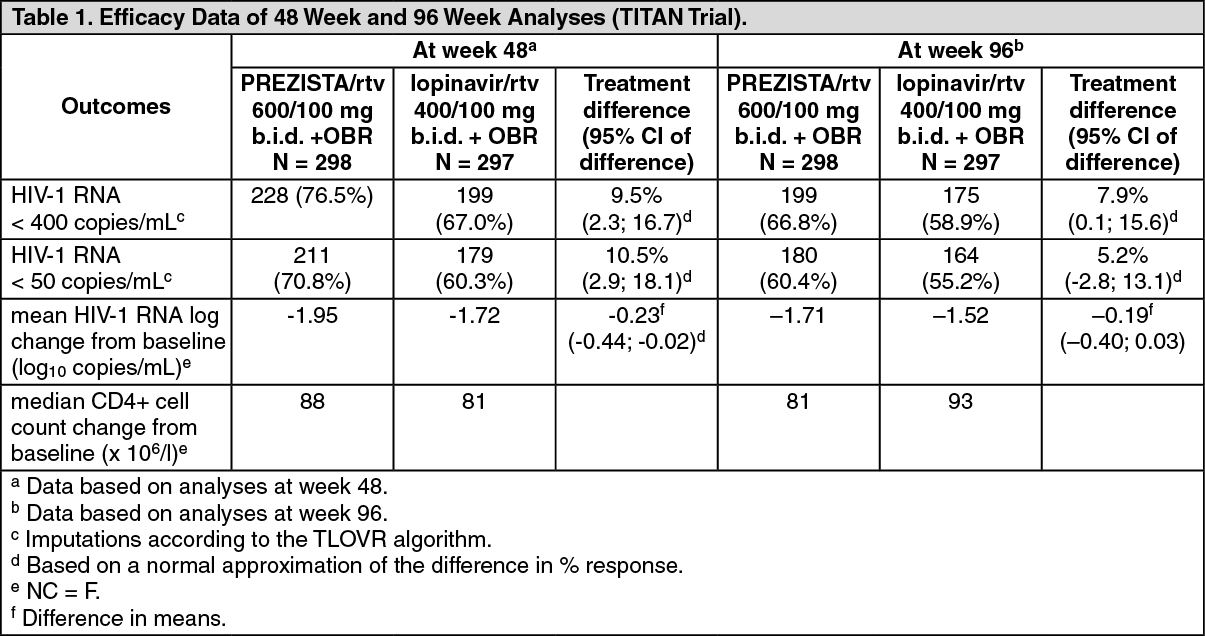
Table 1 as follows shows the efficacy data of the 48 week and 96 week analyses from the TITAN trial. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In the 48 week analysis, virologic response, defined as the percentage of subjects with plasma HIV-1 RNA level < 400 copies/mL, was 76.5% and 67.0% for the PREZISTA/rtv arm and lopinavir/rtv arm, respectively. Non-inferiority in virologic response was demonstrated (p < 0.001) for both ITT and OP populations; furthermore superiority of PREZISTA/rtv over the lopinavir/rtv arm was demonstrated (p = 0.008). 70.8% of patients on PREZISTA/rtv reached less than 50 HIV-1 RNA copies/mL versus 60.3% in the lopinavir/rtv arm.
Analyses of data at 96 weeks of treatment in the TITAN trial demonstrated sustained antiretroviral efficacy and immunological benefit. In the 96 week analysis, virologic response, defined as the percentage of subjects with plasma HIV-1 RNA level < 400 copies/mL, was 66.8% and 58.9% for the PREZISTA/rtv arm and lopinavir/rtv arm, respectively. Non-inferiority in virologic response was demonstrated (p < 0.001) for both ITT and OP populations; furthermore superiority of PREZISTA/rtv over the lopinavir/rtv arm was demonstrated (p = 0.034 for the ITT population and p = 0.033 for the OP population). 60.4% of patients on PREZISTA/rtv reached HIV-1 RNA less than 50 copies/mL versus 55.2% in the lopinavir/rtv arm.
POWER 1 and POWER 2 are randomized, controlled Phase IIb trials in adult patients with a high level of PI resistance, consisting of 2 parts: an initial partially blinded, dose-finding part and a second long term part in which all patients randomized to PREZISTA/rtv received the recommended dose of 600/100 mg b.i.d.
HIV-1 infected patients who were eligible for these trials had plasma HIV-1 RNA > 1000 copies/mL, had prior treatment with PI(s), NNRTI(s) and NRTI(s), had at least 1 primary (i.e., major) PI mutation at screening and were on a stable PI-containing regimen at screening for at least 8 weeks. Randomization was stratified by the number of PI mutations, screening viral load and the use of enfuvirtide.
Demographics and baseline characteristics were balanced between the PREZISTA/rtv arm and the comparator arm. In both trials combined, the 131 patients on PREZISTA/rtv 600/100 mg b.i.d. had a median age of 43 years (range 27 - 73), 89% were male, 81% white, 10% black and 7% hispanic. The mean baseline plasma HIV-1 RNA was 4.61 log
10 copies/mL and the median baseline CD4+ cell count was 153 x 10
6 cells/l (range 3 - 776 x 10
6 cells/l). The median darunavir FC was 4.3. In the PREZISTA/rtv 600/100 mg b.i.d. arm patients had prior exposure to a mean of 4 PIs, 5 NRTIs and 1 NNRTI versus 4 PIs, 6 NRTIs and 1 NNRTI in the comparator arm. Twenty percent of the patients in the PREZISTA/rtv arm had prior use of enfuvirtide versus 17% in the comparator arm.
The virologic response, defined as a decrease in plasma HIV-1 RNA viral load of at least 1.0 log
10 versus baseline, was evaluated in patients receiving PREZISTA/rtv plus an optimized background regimen (OBR) versus a control arm receiving an investigator-selected PI(s) regimen plus an OBR. The OBR consisted of at least 2 NRTIs with or without enfuvirtide (ENF). Based on resistance testing and prior medical history, selected PIs in the control arm included: lopinavir/ritonavir in 36%, (fos)amprenavir in 34%, saquinavir in 35%, and atazanavir in 17%. Twenty-three percent of the control patients used dual-boosted PIs. Approximately 47% of all patients used enfuvirtide and 35% of the use was in patients who were ENF-naive.
POWER 3: additional data on the efficacy of PREZISTA/rtv 600/100 mg b.i.d. have been obtained in treatment-experienced adult patients participating in the non-randomized trial TMC114-C215. At week 48, 334 patients were included in the POWER 3 efficacy analysis who had initiated therapy with PREZISTA/rtv with the recommended dose of 600/100 mg b.i.d. The OBR consisted of at least two NRTIs with or without enfuvirtide. Entry criteria were the same as and baseline characteristics were comparable to those of POWER 1 and POWER 2. The mean baseline plasma HIV-1 RNA was 4.58 log
10 copies/mL and the median CD4+ cell count was 120 x 10
6 cells/l (range 0 - 831 x 10
6 cells/l). The median darunavir FC was 3.2. Patients had a prior exposure to a mean of 5 PIs, 6 NRTIs and 2 NNRTIs, 32% had prior use of enfuvirtide.
Table 2 as follows shows the efficacy data of the 48 week analyses from the pooled POWER 1 and POWER 2 trials as well as from POWER 3. (See Table 2.)
Click on icon to see table/diagram/image
In the pooled POWER 1 and POWER 2 analysis, the proportion of patients in the PREZISTA/rtv (600/100 mg b.i.d.) arm provided superior decreases in log10 viral load from baseline compared to the comparator arm. At week 48, the proportion of patients in the PREZISTA/rtv arm resulted in 62% of patients with a decrease of at least 1.0 log
10 in viral load, compared to 16% in the comparator arm. The proportion of patients with HIV-1 RNA < 50 copies/mL was 45% in the PREZISTA/rtv arm compared to 11% for the comparator arm.
The 48 week efficacy POWER 3 analysis confirmed the viral load reduction and CD4+ increase observed in the POWER 1 and POWER 2 trials. Of the 334 patients included in the week 48 analysis, 59% had a virologic response defined as a decrease of at least 1.0 log
10 in plasma viral load versus baseline and 46% of the patients reached less than 50 HIV-1 RNA copies/mL.
Analyses of data through 96 weeks of treatment in the POWER trials demonstrated sustained antiretroviral efficacy and immunological benefit. Treatment with PREZISTA/rtv (600/100 mg b.i.d.) resulted in 56.5% (POWER 1 and 2) and 52.2% (POWER 3) of patients with a decrease of at least 1 log
10 in HIV-1 RNA from baseline. 38.9% (POWER 1 and 2) and 42.1% (POWER 3) of patients reached an HIV-1 RNA level < 50 copies/mL. At 96 weeks, 49.6% (POWER 1 and 2) and 50.0% (POWER 3) of patients reached less than 400 HIV-1 RNA copies/mL. The mean decrease in HIV-1 RNA level compared to baseline was 1.58 (POWER 1 and 2) and 1.43 (POWER 3) log
10 copies/mL and a mean increase in CD4+ cell count of 133 x 10
6 cells/l (POWER 1 and 2) and 103 x 10
6 cells/l (POWER 3) was observed. Out of the 206 patients who responded with complete viral suppression (< 50 copies/mL) at week 48, 177 patients (86% of the responders at week 48) remained responders at week 96.
In vivo selection of viral resistance during PREZISTA/rtv therapy: In the 96 week analysis of the TITAN trial, the number of virologic failures was lower in the group of subjects receiving PREZISTA/rtv 600/100 mg b.i.d. than in subjects receiving lopinavir/ritonavir 400/100 mg b.i.d. (13.8% vs. 25.6%, respectively). Fewer virologic failures treated with PREZISTA/rtv 600/100 mg b.i.d. than with lopinavir/ritonavir 400/100 mg b.i.d. developed primary (i.e., major) PI mutations (7 vs. 25, respectively) or NRTI RAMs (4 vs. 20, respectively) or lost susceptibility to the PI (3 vs. 17, respectively) or NRTI(s) (4 vs. 20, respectively) used in the treatment regimen.
In a pooled analysis of the POWER and DUET trials, the identified amino acid substitutions that developed on PREZISTA/rtv 600/100 mg b.i.d. in ≥ 20% of the isolates from patients who experienced virological failure by rebound were V32I, I54L, and L89V. Amino acid substitutions that developed in 10 to 20% of the isolates were V11I, I13V, L33F, I50V, and F53L.
In vivo cross-resistance with other HIV protease inhibitors: Of the viruses isolated from subjects receiving PREZISTA/rtv/ 600/100 mg b.i.d. experiencing virologic failures in the TITAN trial, 8% of those susceptible to darunavir at baseline developed decreased susceptibility to darunavir during treatment. In the same group of subjects, 97% to 100% that were susceptible at baseline to amprenavir, atazanavir, indinavir, lopinavir, saquinavir or tipranavir remained susceptible after PREZISTA/rtv treatment.
Of the viruses isolated from patients experiencing virologic failure by rebound from the PREZISTA/rtv 600/100 mg b.i.d. group of the POWER and DUET trials, 85% that were susceptible to darunavir at baseline developed decreased susceptibility to darunavir during treatment. In the same group of patients, 71% of viruses that were susceptible to tipranavir at baseline remained susceptible after treatment. In the POWER trials, patients with resistance to tipranavir (FC > 3) at baseline showed a mean change in viral load at week 24 of -1.38 log
10. Cross-resistance with the other PIs could not be studied in the POWER and DUET trials, since most of the baseline viruses were already resistant to these PIs. Patients with no susceptible PI at baseline (excluding tipranavir) showed a mean change in viral load at week 24 of -1.57 log
10.
Baseline genotype or phenotype and virologic outcome: In a pooled analysis of the 600/100 mg b.i.d groups of the POWER and DUET trials, the presence at baseline of three or more of the mutations V11I, V32I, L33F, I47V, I50V, I54L or M, T74P, L76V, I84V or L89V was associated with a decreased virologic response to PREZISTA/rtv.
In early treatment-experienced patients (TITAN) three or more of these mutations were only found in 4% of the patients at baseline. (See Table 3.)
Click on icon to see table/diagram/image
Baseline darunavir phenotype (shift in susceptibility relative to reference) was shown to be a predictive factor of virologic outcome.
Response rates assessed by baseline darunavir phenotype are shown in the table as follows. The data are provided to give clinicians information on the likelihood of virologic success based on pre-treatment susceptibility to darunavir. (See Table 4.)
Click on icon to see table/diagram/image
In deciding on a new regimen for patients who have failed an antiretroviral regimen, careful consideration should be given to the treatment history and to resistance testing results where available.
Pharmacokinetics: The pharmacokinetic properties of PREZISTA, co-administered with ritonavir, have been evaluated in healthy adult volunteers and in HIV-1 infected patients. Exposure to darunavir was higher in HIV-1 infected patients than in healthy subjects. The increased exposure to darunavir in HIV-1 infected patients compared to healthy subjects may be explained by the higher concentrations of alpha-1-acid glycoprotein (AAG) in HIV-1 infected patients, resulting in higher darunavir binding to plasma AAG and, therefore, higher plasma concentrations.
Darunavir is primarily metabolized by CYP3A. Ritonavir inhibits CYP3A, thereby increasing the plasma concentrations of darunavir considerably.
Absorption: Darunavir was rapidly absorbed following oral administration. Maximum plasma concentration of darunavir in the presence of low-dose ritonavir is generally achieved within 2.5 - 4.0 hours.
The absolute oral bioavailability of a single 600 mg dose of PREZISTA alone was approximately 37% and increased to approximately 82% in the absence of 100 mg b.i.d. ritonavir. The overall pharmacokinetic enhanccement effect by ritonavir was an approximate 14-fold increased in the systemic exposure of darunavir when a single dose of 600 mg PREZISTA was given orally in combination with ritonavir at 100 mg b.i.d. (see Precautions).
When administered without food, the relative bioavailability of PREZISTA in the presence of low-dose ritonavir is 30% lower as compared to intake with food. Therefore, PREZISTA tablets should be taken with ritonavir and with food. The type of food does not affect exposure to darunavir.
Distribution: Darunavir is approximately 95% bound to plasma protein. Darunavir binds primarily to plasma alpha-1-acid-gylcoprotein.
Metabolism: In vitro experiments with human liver microsomes (HLMs) indicate that darunavir primarily undergoes oxidative metabolism. Darunavir is extensively metabolized by the hepatic CYP system and almost exclusively by isozyme CYP3A4. A
14C-darunavir trial in healthy volunteers showed that a majority of the radioactivity in plasma after a single 400/100 mg PREZISTA/rtv dose was due to the parent drug. At least 3 oxidative metabolites of darunavir have been identified in humans; all showed activity that was at least 10-fold less than the activity of darunavir against wildtype HIV.
Elimination: After a 400/100 mg
14C-darunavir/rtv dose, approximately 79.5% and 13.9% of the administered dose of
14C-darunavir could be retrieved in feces and urine, respectively. Unchanged darunavir accounted for approximately 41.2% and 7.7% of the administered dose in feces and urine, respectively. The terminal elimination half-life of darunavir was approximately 15 hours when combined with ritonavir.
The intravenous clearance of darunavir alone (150 mg) and in the presence of low-dose ritonavir was 32.8 l/h and 5.9 l/h, respectively.
Special populations: Elderly (65 years of age and older): Population pharmacokinetic analysis in HIV-infected patients showed that PREZISTA pharmacokinetics are not considerably different in the age range (18-75 years) evaluated in HIV infected patients (see Precautions).
Renal impairment: Results from a mass balance study with
14C-darunavir/rtv showed that approximately 7.7% of the administered dose of darunavir is excreted in the urine as unchanged drug.
Although PREZISTA has not been studied in patients with renal impairment, population pharmacokinetic analysis showed that the pharmacokinetics of PREZISTA were not significantly affected in HIV infected patients with moderate renal impairment (CrCl between 30 - 60 mL/min, n = 20) (see Dosage & Administration and Precautions).
Hepatic impairment: Darunavir is primarily metabolized and eliminated by the liver. In a multiple dose study with PREZISTA co-administered with ritonavir (600/100 mg) twice daily, it was demonstrated that the steady-state pharmacokinetic parameters of darunavir in subjects with mild (Child-Pugh Class A, n = 8) and moderate (Child-Pugh Class B, n = 8) hepatic impairment were comparable with those in healthy subjects. The effect of severe hepatic impairment on the pharmacokinetics of darunavir has not been studied (see Dosage & Administration and Precautions).
Gender: Population pharmacokinetic analysis showed a slightly higher darunavir exposure in HIV infected females compared to males. This difference is not clinically relevant.
Pregnancy and postpartum: The exposure to total darunavir and ritonavir after intake of darunavir/ritonavir 600/100 mg b.i.d as part of an antiretroviral regimen was generally lower during pregnancy compared with postpartum (see Table 5.) However, for unbound (i.e., active) darunavir, the pharmacokinetic parameters were less reduced during pregnancy compared to postpartum due to an increase in the unbound fraction of darunavir during pregnancy compared to postpartum. (See Table 5.)
Click on icon to see table/diagram/image
In women receiving darunavir/ritonavir 600/100 mg b.i.d during the 2nd trimester of pregnancy, mean intra-individual values for total darunavir C
max, AUC
12h and C
min were 28%, 26% and 26% lower, respectively, as compared with postpartum, during the 3rd trimester of pregnancy, total darunavir C
max, AUC
12h and C
min values were 18%, 16% and 2% higher, respectively, as compared with postpartum.
Toxicology: Non-Clinical Information: Carcinogenicity and Mutagenicity: Darunavir was evaluated for carcinogenic potential by oral gavage administration to mice and rats up to 104 weeks. Daily doses of 150, 450 and 1000 mg/kg were administered to mice and doses of 50, 150 and 500 mg/kg were administered to rats. Dose-related increases in the incidences of hepatocellular adenomas and carcinomas were observed in males and females of both species. Thyroid follicular cell adenomas were noted in male rats. Administration of darunavir did not cause a statistically significant increase in the incidence of any other benign or malignant neoplasm in mice or rats. The observed hepatocellular findings in rodents are considered to be of limited relevance to humans. Repeated administration of darunavir to rats caused hepatic microsomal enzyme induction and increased thyroid hormone elimination, which predispose rats, but not humans, to thyroid neoplasms. At the highest tested doses, the systemic exposures (based on AUC) to darunavir were between 0.4- and 0.7-fold (mice) and 0.7- and 1-fold (rats), relative to those observed in humans at the recommended therapeutic doses (600/100 mg twice daily or 800/100 mg once daily).
Darunavir was not mutagenic or genotoxic in a battery of
in vitro and
in vivo assays including bacterial reverse mutation (Ames), chromosomal aberration in human lymphocytes and
in vivo micronucleus test in mice.
Toxicology: Animal toxicology studies have been conducted with darunavir alone, in mice, rats, dogs, and in combination with ritonavir in rats and dogs.
In chronic toxicology studies in rats and dogs, there were only limited effects of treatment with darunavir. In the rat, the key target organs identified were the hematopoietic system, the blood coagulation system, liver, and thyroid, observed at 100 mg/kg/day and above and at exposures below clinical levels. A variable but limited decrease in red blood cell-related parameters was observed, together with increases in activated PTT. The observed liver and thyroid changes were considered to reflect an adaptive response to enzyme induction in the rat rather than an adverse effect. In combination toxicity studies with ritonavir, no additional target organs of toxicity were reported in rats. In the dog, no major toxicity findings or key target organs were identified at doses up to 120 mg/kg/day and exposures equivalent to clinical exposure at the recommended dose.
Reproductive Toxicology: In a study conducted in rats, there were no effects on mating or fertility with PREZISTA treatment up to 1000 mg/kg/day and exposure levels below (AUC - 0.5 fold) of that in humans at the clinically recommended dose. Up to same dose levels, there was no teratogenicity with darunavir in rats and rabbits when treated alone; nor in mice when treated in combination with ritonavir. The exposure levels were lower than those with the recommended clinical dose in humans. In addition, rats treated with combination with ritonavir showed no teratogenicity with the increase in exposure levels which are higher than those with the recommended clinical dose in humans.
Juvenile Toxicity: In a pre and postnatal development assessment in rats, darunavir with and without ritonavir caused a transient reduction in body weight of the offspring during lactation. This was attributed to drug exposure via the milk. No post weaning functions were affected with darunavir alone or in combination with ritonavir. In juvenile rats directly dosed with darunavir (from 20 mg/kg to 1000 mg/kg) up to days 23 to 26 of age, mortality was observed and, in some of the animals, convulsions. Within this age range, exposures in plasma, liver, and brain were dose and age dependent and were considerably greater than those observed in adult rats. These findings were attributed to the ontogeny of the CYP450 liver enzymes involved in the metabolism of darunavir and the immaturity of the blood brain barrier. No treatment related mortalities were noted in juvenile rats dosed at 1000 mg/kg darunavir (single dose) on day 26 of age or at 500 mg/kg (repeated dose) from day 23 to 50 of age, and the exposures and toxicity profile were comparable to those observed in adult rats. Due to uncertainties regarding the rate of development of the human blood brain barrier and liver enzymes, PREZISTA/rtv should not be used in pediatric patients below 3 years of age.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image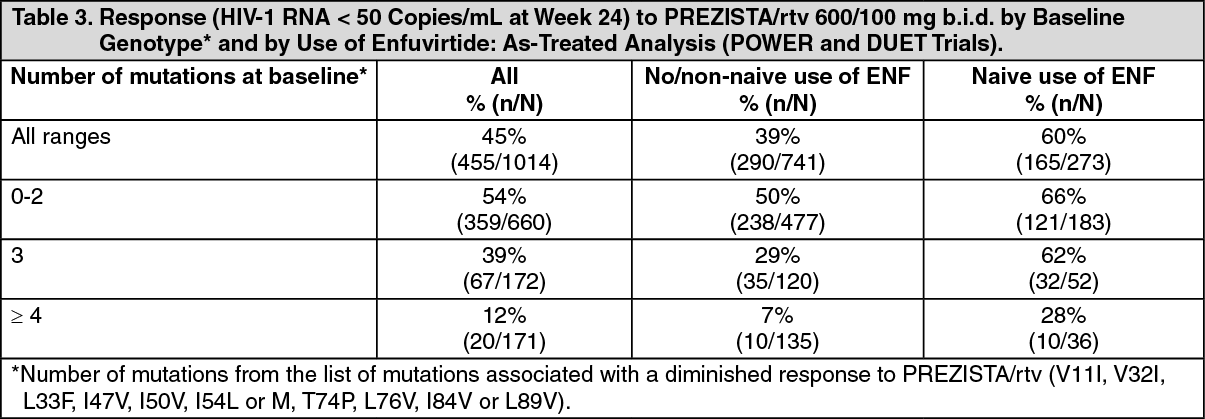 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image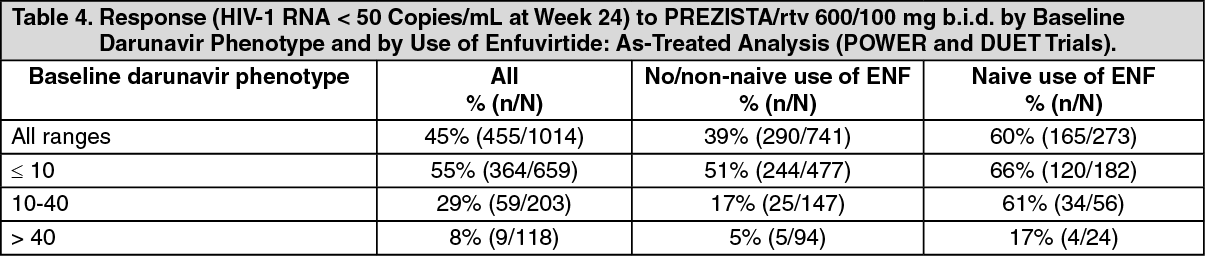 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
