Excipients/Inactive Ingredients: Lactose monohydrate, povidone K25, crospovidone, magnesium stearate, hypromellose, ethylcellulose, macrogol 6000, talc, red ferric oxide (E172), titanium dioxide (E171).
Pharmacotherapeutic group: Imidazoline receptor agonists, moxonidine. ATC Code: C02AC05.
Pharmacology: Pharmacodynamics: In different animal models, moxonidine has been shown to be a potent antihypertensive agent. Available experimental data suggests that the site of the antihypertensive action of moxonidine is the central nervous system (CNS). Within the brainstem, moxonidine has been shown to selectively stimulate imidazoline receptors. These imidazoline-sensitive receptors are concentrated in the rostral ventrolateral medulla, an area critical to the central control of the peripheral sympathetic nervous system. Stimulation of the imidazoline receptors appears to reduce sympathetic activity and lower blood pressure.
Moxonidine distinguishes itself from other sympatholytic antihypertensives by exhibiting only low affinity for known α2-adrenoceptors, as compared to imidazoline receptors. This low affinity to α2-adrenoceptors may account for a low incidence of sedation and dry mouth with moxonidine.
In humans, moxonidine leads to a reduction of systemic vascular resistance and consequently arterial blood pressure. The antihypertensive effect of moxonidine has been demonstrated in double-blind, placebo controlled, randomized studies. Published data show that in hypertensive patients with left ventricular hypertrophy (LVH), for the same blood pressure reduction, the use of an Angiotensin II Antagonist (AIIA) together with moxonidine achieved an improved LVH regression compared to a free combination of a thiazide and a Calcium channel blocker.
In a therapeutic trial of two months' duration, moxonidine improved the insulin sensitivity index by 21% (decreased symptoms related to insulin resistant diabetes) in comparison to placebo in obese and insulin resistant patients with moderate hypertension.
Pharmacokinetics: Absorption: After oral administration of Physiotens, the moxonidine component is rapidly (tmax around 1 h) and almost completely absorbed from the upper gastrointestinal tract. The absolute bioavailability (amount of drug that reaches the blood stream unchanged) is about 88%, indicating no significant first-pass (liver) metabolism. Food intake has no influence on the pharmacokinetics of moxonidine.
Distribution: Plasma protein binding, as determined in vitro, was about 7.2%.
Biotransformation: In pooled human plasma samples, only dehydrogenated moxonidine was identified. The pharmacodynamic activity of dehydrogenated moxonidine is about 1/10 compared to moxonidine.
Elimination: Over a 24-hour period, 78% of the total dose was excreted in urine as parent moxonidine and 13% of the dose was excreted as dehydrogenated moxonidine. Other minor metabolites in urine accounted for approximately 8% of the dose. Less than 1% is eliminated via the faeces. The elimination half-lives of moxonidine and its metabolite are approximately 2.5 h and 5 h, respectively.
Pharmacokinetics in Hypertensive Patients: In hypertensive patients, no relevant pharmacokinetic changes were observed compared to healthy volunteers.
Pharmacokinetics in the Elderly: Age-related changes in pharmacokinetics have been observed and are most likely due to a reduced metabolic activity and/or slightly higher bioavailability (better absorption into the blood stream) in the elderly. However, these pharmacokinetic differences are not considered to be clinically relevant.
Pharmacokinetics in Children: As Physiotens is not recommended for use in children, no pharmacokinetic studies have been performed in this subpopulation.
Pharmacokinetics in Renal Impairment: Elimination of moxonidine is significantly correlated with creatinine clearance. In patients with moderate renal impairment (GFR 30-60 ml/min) steady-state plasma concentrations and terminal half-life are approximately 2 fold and 1.5 fold higher, respectively, compared to hypertensive patients with normal renal function (GFR > 90 ml/min). In patients with severe renal impairment (GFR < 30 ml/min) steady state plasma concentrations and terminal half-life are approximately 3-fold higher. No unexpected drug accumulation after multiple dosing was observed in these patients. In end-stage renal patients (GFR < 10 ml/min) undergoing hemodialysis the AUC (0.2 mg)/plasma concentrations (0.4 mg)and terminal half-lives are 6 fold and 4 fold higher, respectively, compared to hypertensive patients with normal renal function (0.2 mg). In patients with moderate renal impairment (0.2 mg)/in all groups (0.4 mg) maximum moxonidine plasma concentrations are only 1.5-2 fold higher.
In renally impaired patients, the dosage and frequency of dosage should be titrated (0.2 mg)/adjusted (0.4 mg) according to the individual's requirements.
Moxonidine is eliminated to a small extent by hemodialysis.
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction.
Studies in animals have shown embryo-toxicity effects with maternal-toxicity doses.
Studies on reproduction toxicity showed no effect on fertility and no teratogen potential.
Embryotoxicological effects were seen in rat with doses at and above 9 mg/kg/day and in rabbit with doses above 0.7 mg/kg/day. In a peri- and post-natal study on rat influence on development and vitality was noted with doses at and above 3 mg/kg/day.
Physiotens is indicated for the treatment of hypertension (high blood pressure).
The usual starting/initial dose of Physiotens is 0.2 mg daily, with a maximum daily dose of 0.6 mg given as two divided doses. The maximum single dose to be administered is 0.4 mg. Adjustments in daily dose should be individualized to the patient's response.
Physiotens can be taken with or without food.
In patients with moderate to severe renal impairment the starting dose is 0.2 mg daily. If necessary and well tolerated the dose can be increased to 0.4 mg daily in patients with moderate renal impairment and to 0.3 mg daily in patients with severe renal impairment (see Precautions).
In patients undergoing hemodialysis the starting dose is 0.2 mg daily. If necessary and well tolerated the dose can be increased to 0.4 mg daily.
Physiotens is not recommended for use in children and adolescents below 18 years due to lack of data on safety and efficacy.
Symptoms of overdose: In the few cases of overdose that have been reported, a dose of 19.6 mg was ingested acutely without fatality. Signs and symptoms reported included: headache, sedation, somnolence, hypotension, dizziness, asthenia, bradycardia, dry mouth, vomiting, fatigue and upper abdominal pain. In case of a severe overdose close monitoring of especially consciousness disturbances and respiratory depression is recommended.
0.4 mg: In addition, based on a few high dose studies in animals, transient hypertension, tachycardia, and hyperglycaemia may also occur.
Treatment of overdose: No specific antidote is known. In case of hypotension, circulatory support such as fluids and dopamine administration may be considered. Bradycardia may be treated with atropine.
α-Receptor antagonists may diminish or abolish the paradoxical hypertensive effects of a moxonidine overdose.
Physiotens is contraindicated in patients with: Hypersensitivity to moxonidine or to any of the excipients; Sick sinus syndrome; Bradycardia (resting HR < 50 beats/minute); AV-block 2nd and 3rd degree; Cardiac insufficiency.
Cases of varying degrees of AV block have been reported in the post-marketing setting in patients undergoing moxonidine treatment. Based on these case reports, the causative role of moxonidine in delaying atrioventricular conduction cannot be completely ruled out. Therefore, caution is recommended when treating patients with a possible predisposition to developing an AV block. When moxonidine is used in patients with 1st degree AV block, special care should be exercised to avoid bradycardia. Moxonidine must not be used in higher degree AV blocks (see Contraindications).
When moxonidine is used in patients with severe coronary artery disease or unstable angina pectoris, special care should be exercised due to the fact that there is limited experience in this patient population.
Caution is advised in the administration of moxonidine to patients with renal impairment as moxonidine is excreted primarily via the kidney. In these patients careful titration of the dose is recommended, especially at the start of therapy.
Dosing should be initiated with 0.2 mg daily and can be increased to a maximum of 0.4 mg daily for patients with moderate renal impairment (GFR > 30 ml/min but < 60 ml/min) and to a maximum of 0.3 mg daily for patients with severe renal impairment (GFR < 30 ml/min), if clinically indicated and well tolerated.
If moxonidine is used in combination with a β-blocker and both treatments have to be discontinued, the β-blocker should be discontinued first, and then moxonidine after a few days.
So far, no rebound-effect has been observed on the blood pressure after discontinuing the treatment with moxonidine. However, an abrupt discontinuance of the moxonidine treatment is not advisable; instead the dose should be reduced gradually over a period of two weeks.
Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Effects on ability to drive and use machines: 0.4 mg: No studies on the effects of Physiotens on the ability to drive and/or use machinery have been performed. Somnolence and dizziness have been reported. This should be borne in mind when performing these tasks.
Pregnancy: There are no adequate data from use of moxonidine in pregnant woman. Studies in animals have shown embryo-toxicological effects (see Pharmacology: Toxicology: Preclinical safety data under Actions). The potential risk for humans is unknown. Moxonidine should not be used during pregnancy unless clearly necessary.
Lactation: Moxonidine is secreted in breast milk and should therefore not be used during breast feeding. If therapy with moxonidine is considered absolutely necessary, the breast feeding shall be stopped.
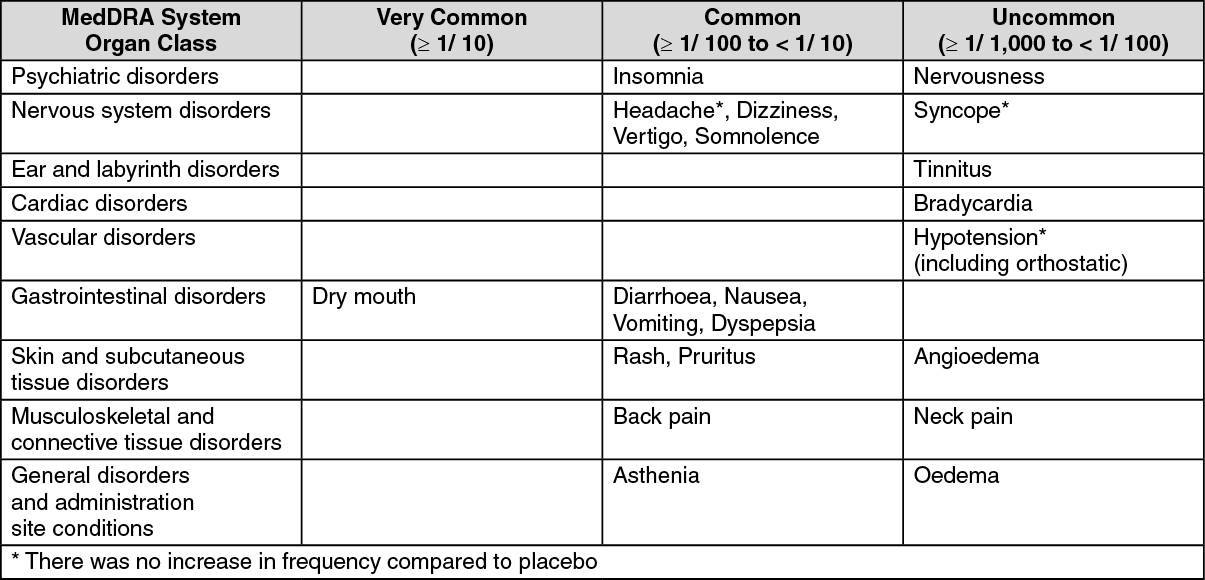
Most frequent side effects reported by those taking moxonidine include dry mouth, dizziness, asthenia and somnolence. These symptoms often decrease after the first few weeks of treatment.
Undesirable Effects by System Organ Class (observed during placebo-controlled clinical trials with n=886 patients exposed to moxonidine resulted in frequencies as follows): (See table.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Concomitant administration moxonidine and other antihypertensive agents result in an additive effect.
Since tricyclic antidepressants may reduce the effectiveness of centrally acting antihypertensive agents, it is not recommended that tricyclic antidepressants be co-administered with moxonidine.
Moxonidine can potentiate the sedative effect of tricyclic anti-depressants (avoid co-prescribing), tranquillizers, alcohol, sedatives and hypnotics.
Moxonidine moderately augmented the impaired performance in cognitive functions in subjects receiving lorazepam. Moxonidine may enhance the sedative effect of benzodiazepines when administered concomitantly.
Moxonidine is excreted through tubular excretion. Interaction with other agents that are excreted through tubular excretion cannot be excluded.
Incompatibilities: Not applicable.
Further information: Medicines should not be disposed of via wastewater or household waste. Ask the pharmacist how to dispose of medicines no longer required. These measures will help to protect the environment.
Do not store above 30°C.
Store in the original package.
Shelf life: 0.2 mg: 2 years.
0.4 mg: 3 years.
C02AC05 - moxonidine ; Belongs to the class of imidazoline receptor agonists, centrally-acting antiadrenergic agents. Used in the treatment of hypertension.
Physiotens FC tab 0.2 mg
28's
Physiotens FC tab 0.4 mg
28's




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
