


 Sign Out
Sign Out




Pharmacology: Pharmacodynamics: Mechanism of action: Nirmatrelvir is a peptidomimetic inhibitor of the coronavirus 3C-like (3CL) protease, including the SARS-CoV-2 3CL protease. Inhibition of the 3CL protease renders the protein incapable of processing polyprotein precursors which leads to the prevention of viral replication. Nirmatrelvir was shown to be a potent inhibitor of SARS-CoV-2 3CL protease (Ki=0.00311 μM or IC50=0.0192 μM) in a biochemical enzymatic assay.
Ritonavir is not active against SARS-CoV-2 3CL protease. Ritonavir inhibits the CYP3A-mediated metabolism of nirmatrelvir, thereby providing increased plasma concentrations of nirmatrelvir.
Antiviral activity: In vitro antiviral activity: Nirmatrelvir exhibited antiviral activity against SARS-CoV-2 infection of dNHBE cells, a primary human lung alveolar epithelial cell line (EC90 value of 181 nM) after Day 3 post‑infection.
In vivo antiviral activity: Nirmatrelvir showed antiviral activity in mouse models with mouse-adapted SARS-CoV-2 infection in BALB/c and 129 mouse strains. Oral administration of nirmatrelvir at 300 mg/kg or 1,000 mg/kg twice daily initiated 4 hours post-inoculation or 1,000 mg/kg twice daily initiated 12 hours post inoculation with SARS-CoV-2 MA10 resulted in reduction of lung viral titres and ameliorated indicators of disease (weight loss and lung pathology) compared to placebo-treated animals.
Antiviral resistance: Because nirmatrelvir is co-administered with low dose ritonavir, there may be a risk of HIV-1 developing resistance to HIV protease inhibitors in individuals with uncontrolled or undiagnosed HIV-1 infection.
Pharmacodynamic effects: Cardiac electrophysiology: No clinically relevant effect of nirmatrelvir on QTcF interval was observed in a double-blind, randomised, placebo-controlled, cross-over study in 10 healthy adults. The model predicted upper bound of 90% confidence interval (CI) for baseline and ritonavir adjusted QTcF estimate was 1.96 ms at approximately 4-fold higher concentration than the mean steady-state peak concentration after a therapeutic dose of nirmatrelvir/ritonavir 300 mg/100 mg.
Clinical efficacy and safety: The efficacy of Paxlovid is based on the interim analysis of EPIC-HR, a Phase 2/3, randomised, double-blind, placebo-controlled study in non-hospitalised symptomatic adult participants with a laboratory confirmed diagnosis of SARS-CoV-2 infection. Participants with COVID-19 symptom onset of ≤ 5 days were included in the study. Participants were randomized (1:1) to receive Paxlovid (nirmatrelvir 300 mg/ritonavir 100 mg) or placebo orally every 12 hours for 5 days. The study excluded individuals with a history of prior COVID-19 infection or vaccination. The primary efficacy endpoint is the proportion of participants with COVID-19 related hospitalisation or death from any cause through Day 28 in the modified intent-to-treat (mITT) analysis set (all treated participants with onset of symptoms ≤ 3 days who had at least one post-baseline visit). Secondary efficacy endpoints included assessments of COVID-19 hospitalisation or death from any cause through Day 28 in the mITT1 analysis set (all treated participants with onset of symptoms ≤ 5 days who had at least one post-baseline visit).
A total of 1,361 participants were randomised to receive either Paxlovid or placebo. At baseline, mean age was 45 years; 52% were male; 63% were White, 5% were Black, 48% were Hispanic or Latino and 20% were Asian; 63% of participants had onset of symptoms ≤ 3 days from initiation of study treatment; 44% of participants were serological negative at baseline. The most frequently reported risk factors were BMI ≥ 25 kg/m2 (1080 [79.4%] participants), tobacco use (501 [36.8%] participants), hypertension (441 [32.4%] participants), age ≥ 60 years (255 [18.7%] participants), and diabetes mellitus (175 [12.9%] participants). Other risk factors were cardiovascular disorder (50 [3.7%] participants), chronic kidney disease (8 [0.6%] participants), chronic lung disease (67 [4.9%] participants), immunosuppression (12 [0.9%] participants), cancer (4 [0.3%] participants), neurodevelopmental disorders (2 [0.1%] participants), HIV infection (1 [<0.1%] participant) and device dependency (5 [0.4%] participants). The mean (SD) baseline viral load was 4.71 log10 copies/mL (2.78); 27% of participants had a baseline viral load of > 10^7 (units); 8.2% of participants either received or were expected to receive COVID-19 therapeutic monoclonal antibody treatment at the time of randomisation and were excluded from the mITT and mITT1 analyses.
The baseline demographic and disease characteristics were balanced between the Paxlovid and placebo groups.
At time of the interim analysis, 389 participants in the Paxlovid group and 385 participants in the placebo group were included in the mITT analysis set. Paxlovid significantly reduced (p<0.0001) the proportion of participants with COVID-19 related hospitalisation or death through Day 28 by 89.1%, compared with placebo, in adult participants with symptom onset ≤ 3 days who were at increased risk of progression to severe disease. No deaths were reported in the Paxlovid group compared with 7 deaths in the placebo group. The proportions of participants who discontinued treatment due to an adverse event were 2.4% in the Paxlovid group and 4.3% in the placebo group.
Similar trends have been observed for the primary efficacy analysis across subgroups of participants. Table 1 presents the results of the primary endpoint in the mITT analysis population and in the subgroups by baseline viral load, serology status or age. (See Table 1.)
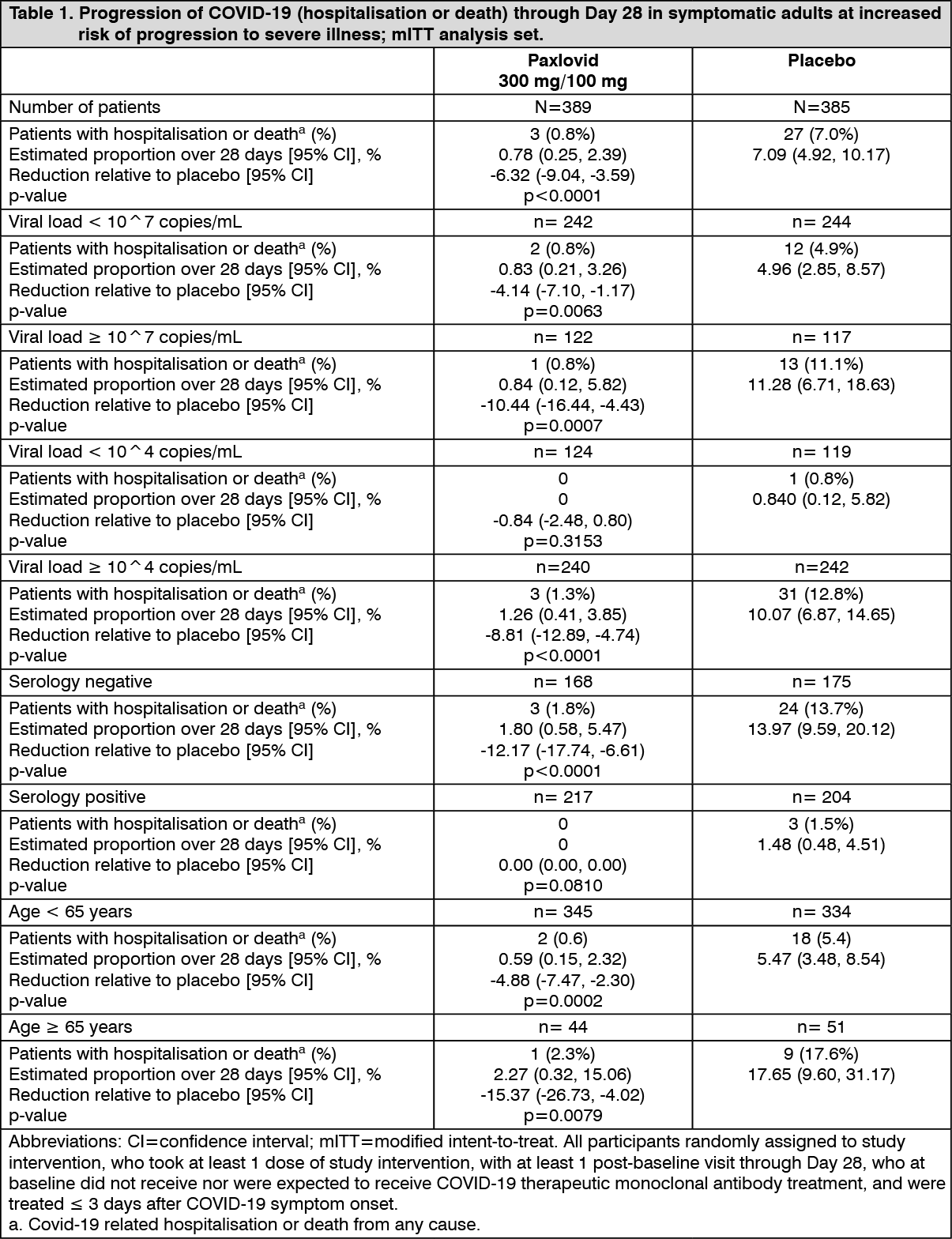 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWhen initiated within 5 days of symptom onset, treatment with Paxlovid also significantly reduced the incidence of hospitalisation or death by 85.2% through Day 28 (Table 2). No deaths were reported in the Paxlovid group compared with 10 deaths in the placebo group. Results of the subgroup analysis for mITT1 were consistent with those for mITT. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAn interim assessment of the effect of Paxlovid on viral load (copies/mL) relative to placebo was conducted. A total of 572 participants with a detectable baseline viral load were included in the interim assessment, and change from baseline to Day 5 (end of treatment) was evaluated. At Day 5, after accounting for baseline viral load level, geographic region, serology status, and symptom onset, the adjusted mean change in viral load (log10 copies/mL) from baseline showed an additional reduction of 0.93 log10 (copies/mL) in the Paxlovid group relative to placebo. The additional viral load reduction from Paxlovid treatment relative to placebo was more apparent among participants who were seronegative or had high viral load level at baseline. Similarly, among participants with symptom onset ≤ 3 days, a reduction of 1.03 log10 (copies/mL) was shown in the Paxlovid group relative to placebo at Day 5. (See Table 3.)
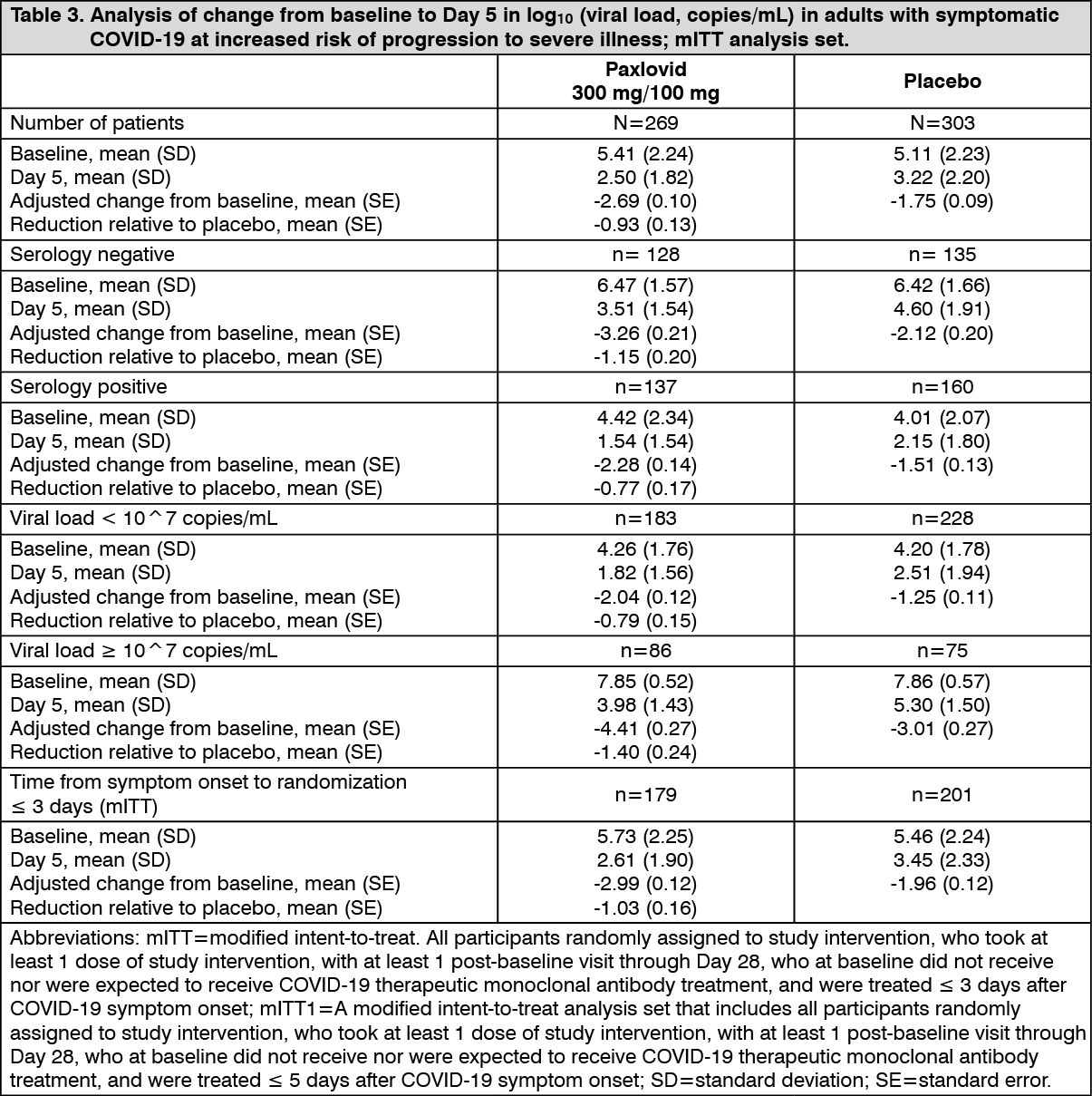 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThis medicinal product has been authorised under a so-called 'conditional approval' scheme. This means that further evidence on this medicinal product is awaited. The Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary.
Paediatric population: The Agency has deferred the obligation to submit the results of studies with Paxlovid in one or more subsets of the paediatric population in the treatment of coronavirus disease 2019 (COVID-19) (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: The pharmacokinetics of nirmatrelvir/ritonavir have been studied in healthy participants.
Ritonavir is administered with nirmatrelvir as a pharmacokinetic enhancer resulting in higher systemic concentrations of nirmatrelvir. In healthy participants in the fasted state, the mean half-life (t½) of a single dose of 150 mg nirmatrelvir administered alone was approximately 2 hours compared to 7 hours after administration of a single dose of 250 mg/100 mg nirmatrelvir/ritonavir thereby supporting a twice-daily administration regimen.
Upon administration of single dose of nirmatrelvir/ritonavir 250 mg/100 mg to healthy participants in the fasted state, the geometric mean (CV%) maximum concentration (Cmax) and area under the plasma concentration-time curve from 0 to the time of last measurement (AUClast) was 2.88 ug/mL (25%) and 27.6 ug*hr/mL (13%), respectively. Upon repeat-dose of nirmatrelvir/ritonavir 75 mg/100 mg, 250 mg/100 mg, and 500 mg/100 mg administered twice daily, the increase in systemic exposure at steady-state appears to be less than dose proportional. Multiple dosing over 10 days achieved steady-state on Day 2 with approximately 2-fold accumulation. Systemic exposures on Day 5 were similar to Day 10 across all doses.
Absorption: Following oral administration of nirmatrelvir/ritonavir 300 mg/100 mg after a single dose, the geometric mean nirmatrelvir (CV%) Cmax and area under the plasma concentration-time curve from 0 to infinity (AUCinf) at steady-state was 2.21 μg/mL (33) and 23.01 μg*hr/mL (23), respectively. The median (range) time to Cmax (Tmax) was 3.00 hrs (1.02-6.00). The arithmetic mean (+SD) terminal elimination half-life was 6.1 (1.8) hours.
Following oral administration of nirmatrelvir/ritonavir 300 mg/100 mg after a single dose, the geometric mean ritonavir (CV%) Cmax and AUCinf was 0.36 μg/mL (46) and 3.60 μg*hr/mL (47), respectively. The median (range) time to Cmax (Tmax) was 3.98 hrs (1.48-4.20). The arithmetic mean (+SD) terminal elimination half-life was 6.1 (2.2) hours.
Effect of food on oral absorption: Dosing with a high fat meal modestly increased the exposure of nirmatrelvir (approximately 15% increase in mean Cmax and 1.6% increase in mean AUClast) relative to fasting conditions following administration of a suspension formulation of nirmatrelvir co-administered with ritonavir tablets.
Distribution: The protein binding of nirmatrelvir in human plasma is approximately 69%.
The protein binding of ritonavir in human plasma is approximately 98-99%.
Biotransformation: In vitro studies assessing nirmatrelvir without concomitant ritonavir suggest that nirmatrelvir is primarily metabolised by CYP3A4. Nirmatrelvir does not reversibly inhibit CYP2D6, CYP2C9, CYP2C19, CYP2C8, or CYP1A2 in vitro at clinically relevant concentrations. In vitro study results showed nirmatrelvir may be inducer of CYP3A4, CYP2B6, CYP2C8, and CYP2C9. The clinical relevance is unknown. Based on in vitro data, nirmatrelvir has a low potential to inhibit BCRP, MATE2K, OAT1, OAT3, OATP1B3 and OCT2. There is a potential for nirmatrelvir to inhibit MDR1, MATE1, OCT1 and OATP1B1 at clinically relevant concentrations. Administration of nirmatrelvir with ritonavir inhibits the metabolism of nirmatrelvir. In plasma, the only drug-related entity observed was unchanged nirmatrelvir. Minor oxidative metabolites were observed in the faeces and urine.
In vitro studies utilising human liver microsomes have demonstrated that cytochrome P450 3A (CYP3A) is the major isoform involved in ritonavir metabolism, although CYP2D6 also contributes to the formation of oxidation metabolite M-2.
Low doses of ritonavir have shown profound effects on the pharmacokinetics of other protease inhibitors (and other products metabolised by CYP3A4) and other protease inhibitors may influence the pharmacokinetics of ritonavir.
Ritonavir has a high affinity for several cytochrome P450 (CYP) isoforms and may inhibit oxidation with the following ranked order: CYP3A4 > CYP2D6. Ritonavir also has a high affinity for P-glycoprotein (P-gp) and may inhibit this transporter. Ritonavir may induce glucuronidation and oxidation by CYP1A2, CYP2C8, CYP2C9 and CYP2C19 thereby increasing the biotransformation of some medicinal products metabolised by these pathways and may result in decreased systemic exposure to such medicinal products, which could decrease or shorten their therapeutic effect.
Elimination: The primary route of elimination of nirmatrelvir when administered with ritonavir was renal excretion of intact drug. Approximately 49.6% and 35.3% of the administered dose of nirmatrelvir 300 mg was recovered in urine and faeces, respectively. Nirmatrelvir was the predominant drug-related entity with small amounts of metabolites arising from hydrolysis reactions in excreta. In plasma, the only drug-related entity quantifiable was unchanged nirmatrelvir.
Human studies with radiolabelled ritonavir demonstrated that the elimination of ritonavir was primarily via the hepatobiliary system; approximately 86% of radiolabel was recovered from stool, part of which is expected to be unabsorbed ritonavir.
Specific populations: The pharmacokinetics of nirmatrelvir/ritonavir based on age and gender have not been evaluated.
Racial or ethnic groups: Systemic exposure in Japanese participants was numerically lower but not clinically meaningfully different than those in Western participants.
Patients with renal impairment: Compared to healthy controls with no renal impairment, the Cmax and AUC of nirmatrelvir in patients with mild renal impairment was 30% and 24% higher, in patients with moderate renal impairment was 38% and 87% higher, and in patients with severe renal impairment was 48% and 204% higher, respectively.
Patients with hepatic impairment: Compared to healthy controls with no hepatic impairment, the pharmacokinetics of nirmatrelvir in subjects with moderate hepatic impairment was not significantly different.
Interaction studies conducted with nirmatrelvir/ritonavir: CYP3A4 was the major contributor to the oxidative metabolism of nirmatrelvir, when nirmatrelvir was tested alone in human liver microsomes. Ritonavir is an inhibitor of CYP3A and increases plasma concentrations of nirmatrelvir and other drugs that are primarily metabolised by CYP3A. Despite being coadministered with ritonavir as a pharmacokinetic enhancer, there is potential for strong inhibitors and inducers to alter the pharmacokinetics of nirmatrelvir.
The effects of co-administration of Paxlovid with itraconazole (CYP3A inhibitor) and carbamazepine (CYP3A inducer) on the nirmatrelvir AUC and Cmax are summarised in Table 4 (effect of other medicinal products on nirmatrelvir). (See Table 4.)
Click on icon to see table/diagram/imagePreclinical safety data: Toxicology: Repeat-dose toxicity studies up to 1 month duration of nirmatrelvir in rats and monkeys resulted in no adverse findings.
Repeat-dose toxicity studies of ritonavir in animals identified major target organs as the liver, retina, thyroid gland and kidney. Hepatic changes involved hepatocellular, biliary and phagocytic elements and were accompanied by increases in hepatic enzymes. Hyperplasia of the retinal pigment epithelium and retinal degeneration have been seen in all of the rodent studies conducted with ritonavir, but have not been seen in dogs. Ultrastructural evidence suggests that these retinal changes may be secondary to phospholipidosis. However, clinical trials revealed no evidence of medicinal product-induced ocular changes in humans. All thyroid changes were reversible upon discontinuation of ritonavir. Clinical investigation in humans has revealed no clinically significant alteration in thyroid function tests.
Renal changes including tubular degeneration, chronic inflammation and proteinuria were noted in rats and are felt to be attributable to species-specific spontaneous disease. Furthermore, no clinically significant renal abnormalities were noted in clinical trials.
Carcinogenesis: Nirmatrelvir has not been evaluated for the potential to cause carcinogenicity.
Long-term carcinogenicity studies of ritonavir in mice and rats revealed tumorigenic potential specific for these species, but are regarded as of no relevance for humans.
Mutagenesis: Paxlovid has not been evaluated for the potential to cause mutagenicity.
Nirmatrelvir was not genotoxic in a battery of assays, including bacterial mutagenicity, chromosome aberration using human lymphoblastoid TK6 cells and in vivo rat micronucleus assays.
Ritonavir was found to be negative for mutagenic or clastogenic activity in a battery of in vitro and in vivo assays including the Ames bacterial reverse mutation assay using S. typhimurium and E. coli, the mouse lymphoma assay, the mouse micronucleus test and chromosomal aberration assays in human lymphocytes.
Reproductive toxicity: Nirmatrelvir: In a fertility and early embryonic development study, nirmatrelvir was administered to male and female rats by oral gavage at doses of 60, 200, or 1,000 mg/kg/day once daily beginning 14 days prior to mating, throughout the mating phase, and continued through Gestation Day (GD) 6 for females and for a total of 32 doses for males. There were no effects on fertility, reproductive performance, or early embryonic development at doses up to 1,000 mg/kg/day representing 12x/4.3x based on the predicted human Cmax/AUC24 at a twice-daily dose of 300 mg/100 mg nirmatrelvir/ritonavir.
The potential embryo-foetal toxicity of nirmatrelvir was evaluated in the definitive rat and rabbit studies at doses up to 1,000 mg/kg/day. There was no nirmatrelvir-related effect in any of the parameters in the rat embryo-foetal development (EFD) study up to the highest dose of 1,000 mg/kg/day (exposure margin of 16x/7.8x based on total Cmax/AUC24 over the predicted human exposures at a dose of 300 mg/100 mg nirmatrelvir/ritonavir twice daily). In the rabbit EFD study, there was no nirmatrelvir-related effect on foetal morphology or embryo-foetal viability up to the highest dose of 1,000 mg/kg/day (exposure margin of 24x/10x based on total Cmax/AUC24), however adverse nirmatrelvir-related lower foetal body weights (0.91x control) were observed at 1,000 mg/kg/day in the presence of non-adverse, low magnitude effects on maternal body weight change and food consumption at this dose. Growth delay is likely reversible following cessation of exposure in human, and it was not present at the intermediate dose (10x/2.8x Cmax/AUC24 over the predicted clinical exposure). There were no nirmatrelvir-related severe manifestations of developmental toxicity (malformations and embryo-foetal lethality) at the highest dose tested, 1,000 mg/kg/day.
Ritonavir: Ritonavir produced no effects on fertility in rats.
Ritonavir was administered orally to pregnant rats (at 0, 15, 35, and 75 mg/kg/day) and rabbits (at 0, 25, 50, and 110 mg/kg/day) during organogenesis (on GD 6 through 17 and 6 through 19, respectively). No evidence of teratogenicity due to ritonavir was observed in rats and rabbits. Increased incidences of early resorptions, ossification delays and developmental variations, as well as decreased foetal body weights were observed in the rat in the presence of maternal toxicity. A slight increase in the incidence of cryptorchidism was also noted in rats (at a maternally toxic dose). In the rabbit, resorptions, decreased litter size and decreased foetal weights were observed in the presence of maternal toxicity. In pre- and post-natal development study in rats, administration 0, 15, 35, and 60 mg/kg/day ritonavir from GD 6 through Post-natal Day 20 resulted in no developmental toxicity.