


 Sign Out
Sign Out

Evidence from several pre-clinical models inform that the pharmacological activity of gabapentin may be mediated via binding to α2δ through a reduction in release of excitatory neurotransmitters in regions of the central nervous system. Such activity may underlie gabapentin's anti-seizure activity. The relevance of these actions of gabapentin to the anticonvulsant effects in humans remains to be established.
Gabapentin also displays efficacy in several pre-clinical animal pain models. Specific binding of gabapentin to the α2δ subunit is proposed to result in several different actions that may be responsible for analgesic activity in animal models. The analgesic activities of gabapentin may occur in the spinal cord as well as at higher brain centers through interactions with descending pain inhibitory pathways. The relevance of these pre-clinical properties to clinical action in humans is unknown.
Pharmacokinetics: Gabapentin bioavailability is not dose-proportional. That is, as the dose is increased, bioavailability decreases. Following oral administration, peak plasma gabapentin concentrations are observed within 2 to 3 hours. Absolute bioavailability of gabapentin capsules is approximately 60%. Food, including a high-fat diet, has no effect on gabapentin pharmacokinetics.
Gabapentin elimination from plasma is best described by linear pharmacokinetics.
The elimination half-life of gabapentin is independent of dose and averages 5 to 7 hours.
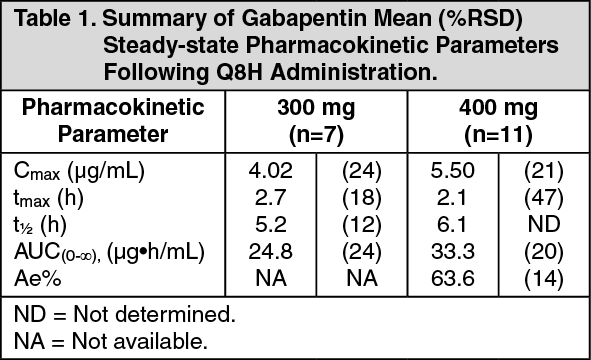
Gabapentin pharmacokinetics are not affected by repeated administration, and steady-state plasma concentrations are predictable from single-dose data. Although plasma gabapentin concentrations were generally between 2 μg/mL and 20 μg/mL in clinical studies, such concentrations were not predictive of safety or efficacy. Plasma gabapentin concentrations are dose proportional at doses of 300 mg or 400 mg given every 8 hours. Pharmacokinetic parameters are given in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageGabapentin is not bound to plasma proteins and has a volume of distribution equal to 57.7 L. In patients with epilepsy, gabapentin concentrations in the Cerebrospinal fluid (CSF) are approximately 20% of corresponding steady-state trough plasma concentrations. Gabapentin is eliminated solely by renal excretion. There is no evidence of metabolism in man. Gabapentin does not induce hepatic mixed function oxidase enzymes responsible for drug metabolism.
In elderly patients, and in patients with impaired renal function, gabapentin plasma clearance is reduced. Gabapentin elimination-rate constant, plasma clearance, and renal clearance are directly proportional to creatinine clearance.
Gabapentin is removed from plasma by hemodialysis. Dose adjustment in patients with compromised renal function or in those undergoing hemodialysis is recommended (see Dose adjustment in impaired renal function in patients with neuropathic pain or epilepsy and Dose adjustment in patients undergoing hemodialysis under Dosage & Administration).
Gabapentin pharmacokinetics in children were determined in 24 healthy subjects between the ages of 4 and 12 years. In general, gabapentin plasma concentrations in children are similar to those in adults.
In a pharmacokinetic study in 24 healthy infants and children, pediatric subjects between 1 and 48 months of age achieved approximately 30% lower exposure (AUC) than that observed in pediatric subjects older than 5 years of age; Cmax was lower and the clearance per body weight was higher in infants and younger children.
Toxicology: Preclinical Safety Data: Carcinogenesis: Gabapentin was given in the diet to mice at 200, 600, and 2000 mg/kg/day and to rats at 250, 1000, and 2000 mg/kg/day for 2 years. A statistically significant increase in the incidence of pancreatic acinar cell tumors was found only in male rats at the highest dose. Peak plasma drug concentrations in rats at 2000 mg/kg/day were 10 times higher than plasma concentrations in humans given at 3600 mg/day. The pancreatic acinar cell tumors in male rats were low-grade malignancies, which did not affect survival, did not metastasize or invade surrounding tissue, and were similar to those seen in concurrent controls. The relevance of these pancreatic acinar cell tumors in male rats to carcinogenic risk in humans is unclear.
Mutagenesis: Gabapentin demonstrated no genotoxic potential. It was not mutagenic in vitro in standard assays using bacterial or mammalian cells. Gabapentin did not induce structural chromosome aberrations in mammalian cells in vitro or in vivo, and did not induce micronucleus formation in the bone marrow of hamsters.
Impairment of fertility: No adverse effects on fertility or reproduction were observed in rats at doses up to 2000 mg/kg (approximately 5 times the maximum daily human dose, on a mg/m2 basis).
Teratogenesis: Gabapentin did not increase the incidence of malformations, compared to controls, in the offsprings of mice, rats, or rabbits at doses up to 50, 30, and 25 times, respectively, the daily human dose of 3600 mg (4, 5 or 8 times, respectively, the human daily dose, on a mg/m2 basis).
Gabapentin induced delayed ossification in the skull, vertebrae, forelimbs, and hindlimbs in rodents, indicative of fetal growth retardation. These effects occurred when pregnant mice received oral doses of 1000 or 3000 mg/kg/day during organogenesis and in rats given 2000 mg/kg/day prior to and during mating and throughout gestation. These doses are approximately 1 to 5 times the human dose of 3600 mg, on a mg/m2 basis.
No effects were observed in pregnant mice given 500 mg/kg/day (approximately half of the daily human dose, on a mg/m2 basis).
An increased incidence of hydroureter and/or hydronephrosis was observed in rats given 2000 mg/kg/day in a fertility and general reproduction study; 1500 mg/kg/day in a teratology study; and 500, 1000, and 2000 mg/kg/day in a perinatal and postnatal study. The significance of these findings is unknown, but they have been associated with delayed development. These doses are also approximately 1 to 5 times the human dose of 3600 mg on a mg/m2 basis.
In a teratology study in rabbits, an increased incidence of post-implantation fetal loss occurred in female rabbits given 60, 300, and 1500 mg/kg/day during organogenesis. These doses are approximately 1/4 to 8 times the daily human dose of 3600 mg, on a mg/m2 basis.