Pharmacotherapeutic group: immunostimulants, colony stimulating factor.
ATC Code: L03AA13
.
Pharmacology: Pharmacodynamics: Human granulocyte-colony stimulating factor (G-CSF) is a glycoprotein, which regulates the production and release of neutrophils from the bone marrow. Pegfilgrastim is a covalent conjugate of recombinant human G-CSF (r-metHuG-CSF) with a single 20 kd polyethylene glycol (PEG) molecule. Pegfilgrastim is a sustained duration form of filgrastim due to decreased renal clearance. Pegfilgrastim and filgrastim have been shown to have identical modes of action, causing a marked increase in peripheral blood neutrophil counts within 24 hours, with minor increases in monocytes and/or lymphocytes. Similarly to filgrastim, neutrophils produced in response to pegfilgrastim show normal or enhanced function as demonstrated by tests of chemotactic and phagocytic function. As with other haematopoietic growth factors, G-CSF has shown
in vitro stimulating properties on human endothelial cells. G-CSF can promote growth of myeloid cells, including malignant cells,
in vitro and similar effects may be seen on some non-myeloid cells
in vitro.
In two randomised, double-blind, pivotal studies in patients with high-risk stage II - IV breast cancer undergoing myelosuppressive chemotherapy consisting of doxorubicin and docetaxel, use of pegfilgrastim, as a single once per cycle dose, reduced the duration of neutropenia and the incidence of febrile neutropenia similarly to that observed with daily administrations of filgrastim (a median of 11 daily administrations). In the absence of growth factor support, this regimen has been reported to result in a mean duration of grade 4 neutropenia of 5 to 7 days, and a 30-40% incidence of febrile neutropenia. In one study (n = 157), which used a 6 mg fixed dose of pegfilgrastim the mean duration of grade 4 neutropenia for the pegfilgrastim group was 1.8 days compared with 1.6 days in the filgrastim group (difference 0.23 days, 95% CI -0.15, 0.63). Over the entire study, the rate of febrile neutropenia was 13% of pegfilgrastim-treated patients compared with 20% of filgrastim-treated patients (difference -7%, 95% CI of -19%, 5%). In a second study (n = 310), which used a weight-adjusted dose (100 μg/kg), the mean duration of grade 4 neutropenia for the pegfilgrastim group was 1.7 days, compared with 1.8 days in the filgrastim group (difference 0.03 days, 95% CI -0.36, 0.30). The overall rate of febrile neutropenia was 9% of patients treated with pegfilgrastim and 18% of patients treated with filgrastim (difference 9%, 95% CI of -16.8%, -1.1%).
In a placebo-controlled study, double-blind study in patients with breast cancer, the effect of pegfilgrastim on the incidence of febrile neutropenia was evaluated following administration of a chemotherapy regimen associated with a febrile neutropenia rate of 10-20% (docetaxel 100 mg/m
2 every 3 weeks for 4 cycles). Nine hundred and twenty eight patients were randomised to receive either a single dose of pegfilgrastim or placebo approximately 24 hours (Day 2) after chemotherapy in each cycle. The incidence of febrile neutropenia was lower for patients randomised to receive pegfilgrastim compared with placebo (1% versus 17%, p ≤ 0.001). The incidence of hospitalisations and IV anti-infective use associated with a clinical diagnosis of febrile neutropenia was lower in the pegfilgrastim group compared with placebo (1% versus 14%, p < 0.001; and 2% versus 10%, p < 0.001).
A small (n = 83), phase II, randomised, double-blind study in patients receiving chemotherapy for
de novo acute myeloid leukaemia compared pegfilgrastim (single dose of 6 mg) with filgrastim, administered during induction chemotherapy. Median time to recovery from severe neutropenia was estimated as 22 days in both treatment groups. Long term outcome was not studied (see Precautions).
In a phase II (n = 37) multicentre, randomised, open-label study of paediatric sarcoma patients receiving 100 μg/kg pegfilgrastim following cycle 1 of vincristine, doxorubicin and cyclophosphamide (VAdriaC/IE) chemotherapy, a longer duration of severe neutropenia (neutrophils < 0.5 x 10
9) was observed in younger children aged 0-5 years (8.9 days) compared to older children aged 6-11 years and 12-21 years (6 days and 3.7 days, respectively) and adults. Additionally a higher incidence of febrile neutropenia was observed in younger children aged 0-5 years (75%) compared to older children aged 6 11 years and 12-21 years (70% and 33%, respectively) and adults (see Adverse Reactions and Pharmacokinetics as follows).
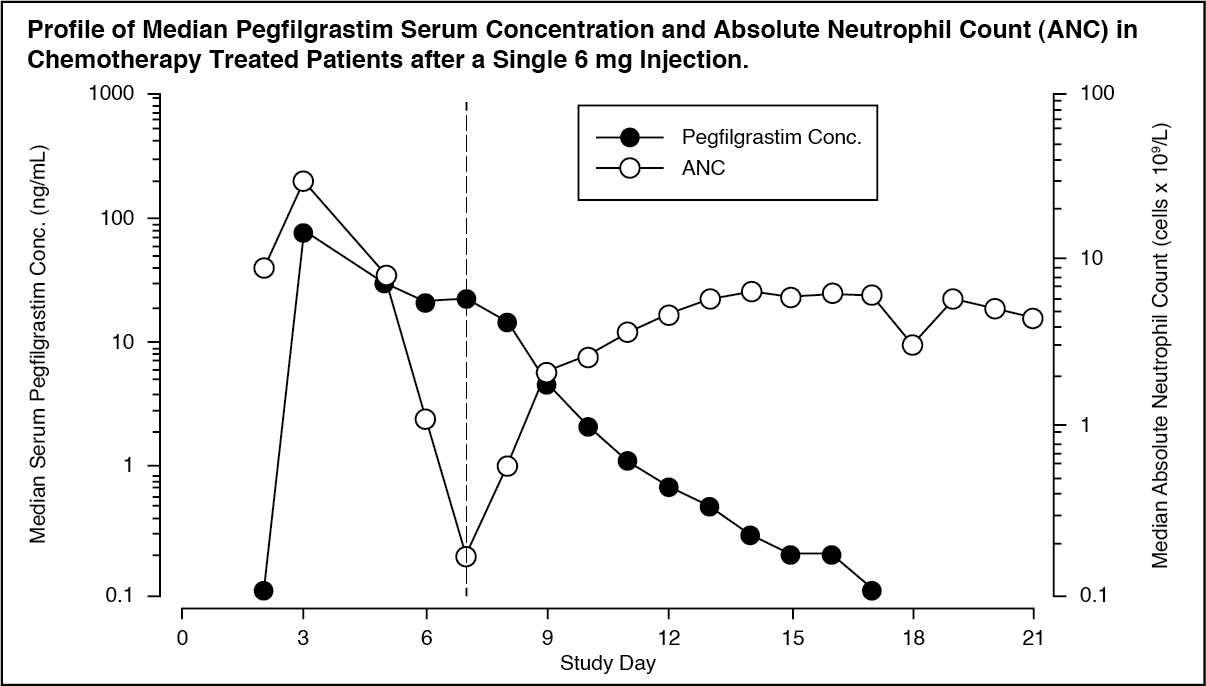
Pharmacokinetics: After a single subcutaneous dose of pegfilgrastim, the peak serum concentration of pegfilgrastim occurs at 16 to 120 hours after dosing and serum concentrations of pegfilgrastim are maintained during the period of neutropenia after myelosuppressive chemotherapy. The elimination of pegfilgrastim is non linear with respect to dose; serum clearance of pegfilgrastim decreases with increasing dose. Pegfilgrastim appears to be mainly eliminated by neutrophil-mediated clearance, which becomes saturated at higher doses. Consistent with a self regulating clearance mechanism, the serum concentration of pegfilgrastim declines rapidly at the onset of neutrophil recovery (see figure).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Due to the neutrophil-mediated clearance mechanism, the pharmacokinetics of pegfilgrastim is not expected to be affected by renal or hepatic impairment. In an open-label, single dose study (n = 31) various stages of renal impairment, including end-stage renal disease, had no impact on the pharmacokinetics of pegfilgrastim.
Elderly people: Limited data indicate that the pharmacokinetics of pegfilgrastim in elderly subjects (> 65 years) is similar to that in adults.
Paediatric population: The pharmacokinetics of pegfilgrastim were studied in 37 paediatric patients with sarcoma, who received 100 μg/kg pegfilgrastim after the completion of VAdriaC/IE chemotherapy. The youngest age group (0-5 years) had a higher mean exposure to pegfilgrastim (AUC) (± Standard Deviation) (47.9 ± 22.5 μg·hr/ml) than older children aged 6-11 years and 12-21 years (22.0 ± 13.1 μg·hr/ml and 29.3 ± 23.2 μg·hr/ml, respectively) (see Pharmacodynamics as previously mentioned). With the exception of the youngest age group (0-5 years), the mean AUC in paediatric subjects appeared similar to that for adult patients with high-risk stage II-IV breast cancer and receiving 100 μg/kg pegfilgrastim after the completion of doxorubicin/docetaxel (see Adverse Reactions and Pharmacodynamics as previously mentioned).
Toxicology: Preclinical safety data: Preclinical data from conventional studies of repeated dose toxicity revealed the expected pharmacological effects including increases in leukocyte count, myeloid hyperplasia in bone marrow, extramedullary haematopoiesis and splenic enlargement.
There were no adverse effects observed in offspring from pregnant rats given pegfilgrastim subcutaneously, but in rabbits pegfilgrastim has been shown to cause embryo/foetal toxicity (embryo loss) at cumulative doses approximately 4 times the recommended human dose, which were not seen when pregnant rabbits were exposed to the recommended human dose. In rat studies, it was shown that pegfilgrastim may cross the placenta. Studies in rats indicated that reproductive performance, fertility, oestrous cycling, days between pairing and coitus, and intrauterine survival were unaffected by pegfilgrastim given subcutaneously. The relevance of these findings for humans is not known.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
