Pharmacotherapeutic group: Other antithrombotic agents.
ATC code: B01AF03.
Pharmacology: Pharmacodynamics: Mechanism of action: Edoxaban is a highly selective, direct and reversible inhibitor of factor Xa, the serine protease located in the final common pathway of the coagulation cascade. Edoxaban inhibits free factor Xa, and prothrombinase activity. Inhibition of factor Xa in the coagulation cascade reduces thrombin generation, prolongs clotting time and reduces the risk of thrombus formation.
Pharmacodynamic effects: Edoxaban produces rapid onset of pharmacodynamic effects within 1 - 2 hours, which corresponds with peak edoxaban exposure (C
max). The pharmacodynamic effects measured by anti-factor Xa assay are predictable and correlate with the dose and the concentration of edoxaban. As a result of FXa inhibition, edoxaban also prolongs clotting time in tests such as prothrombin time (PT), and activated partial thromboplastin time (aPTT). Changes observed in these clotting tests are expected at the therapeutic dose, however, these changes are small, subject to a high degree of variability, and not useful in monitoring the anticoagulation effect of edoxaban.
Effects of coagulation markers when switching from rivaroxaban, dabigatran, or apixaban to edoxaban: In clinical pharmacology studies, healthy subjects received rivaroxaban 20 mg once daily, dabigatran 150 mg twice daily, or apixaban 5 mg twice daily, followed by a single dose of edoxaban 60 mg on Day 4. The effect on prothrombin time (PT) and other coagulation biomarkers (e.g. anti-FXa, aPTT) was measured. Following the switch to edoxaban on Day 4 the PT was equivalent to Day 3 of rivaroxaban and apixaban. For dabigatran higher aPTT activity was observed after edoxaban administration with prior dabigatran treatment compared to that after treatment with edoxaban alone. This is considered to be due to the carry-over effect of dabigatran treatment, however, this did not lead to a prolongation of bleeding time.
Based on these data, when switching from these anticoagulants to edoxaban, the first dose of edoxaban can be initiated at the time of the next scheduled dose of the previous anticoagulant (see Dosage & Administration).
Clinical efficacy and safety: Prevention of stroke and systemic embolism: The edoxaban clinical programme for atrial fibrillation was designed to demonstrate the efficacy and safety of two dose groups of edoxaban compared to warfarin for the prevention of stroke and systemic embolism in subjects with nonvalvular atrial fibrillation and at moderate to high risk of stroke and systemic embolic events (SEE).
In the pivotal ENGAGE AF-TIMI 48 study (an event-driven, Phase 3, multi-centre, randomised, double-blind double-dummy parallel-group study), 21,105 subjects, with a mean CHADS
2 score of 2.8, were randomised to either edoxaban 30 mg once daily treatment group, or edoxaban 60 mg once daily treatment group or warfarin. Subjects in both edoxaban treatment groups had their dose halved if one or more of the following clinical factors were present: moderate renal impairment (CrCL 30 - 50 mL/min), low body weight (≤ 60 kg) or concomitant use of specific P-gp inhibitors (verapamil, quinidine, dronedarone).
The primary efficacy endpoint was the composite of stroke and SEE. Secondary efficacy endpoints included: Composite of stroke, SEE, and cardiovascular (CV) mortality; major adverse cardiovascular event (MACE), which is the composite of non-fatal MI, non-fatal stroke, non-fatal SEE, and death due to CV cause or bleeding; composite of stroke, SEE, and all-cause mortality.
The median study drug exposure for both the edoxaban 60 mg and 30 mg treatment groups was 2.5 years. The median study follow-up for both the edoxaban 60 mg and 30 mg treatment groups was 2.8 years. The median subject-year exposure was 15,471, and 15,840 for the 60 mg and 30 mg treatment groups, respectively; and the median subject-year follow-up was 19,191 and 19,216 for the 60 mg and 30 mg treatment groups, respectively.
In the warfarin group, the median TTR (time in therapeutic range, INR 2.0 to 3.0) was 68.4%.
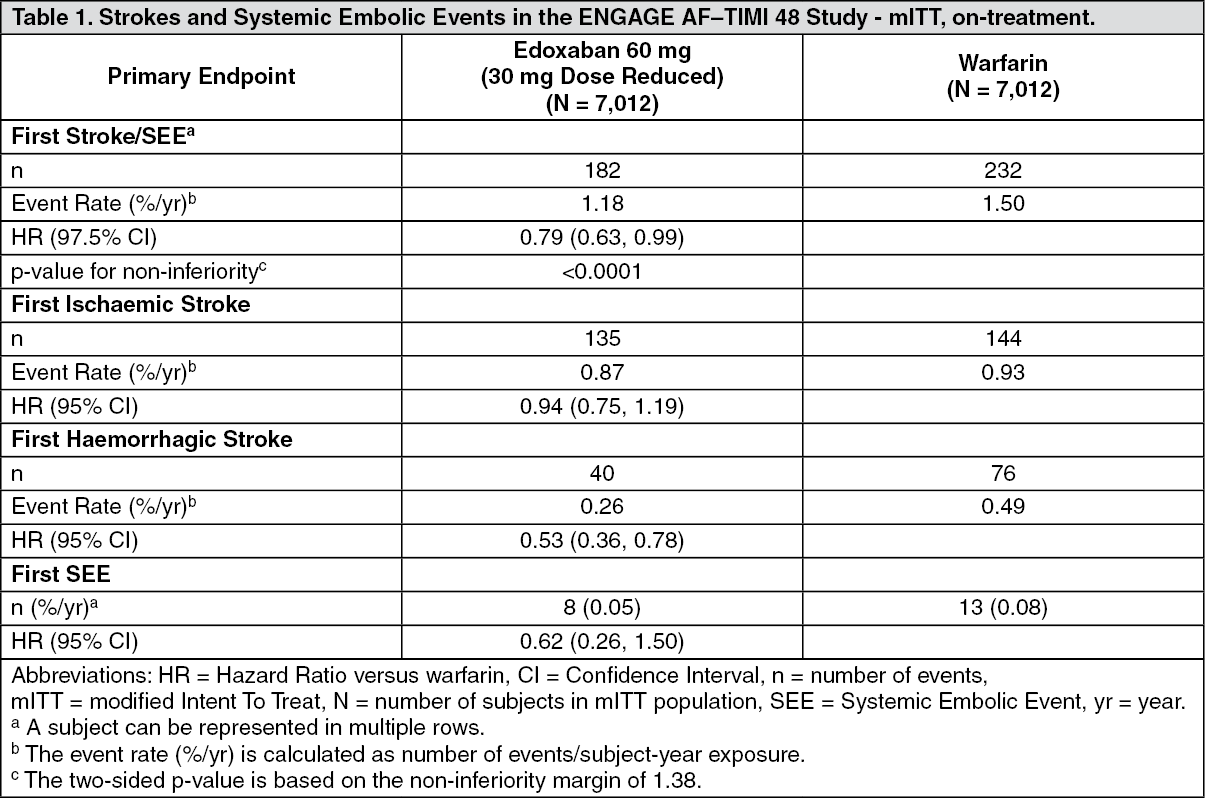
The main analysis of efficacy was aimed to show the non-inferiority of edoxaban versus warfarin on first stroke or SEE that occurred during treatment or within 3 days from the last dose taken in the modified intention-to-treat (mITT) population. Edoxaban 60 mg was non-inferior to warfarin for the primary efficacy endpoint of stroke or SEE (upper limit of the 97.5% CI of the HR was below the pre-specified non-inferiority margin of 1.38) (Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
During the overall study period in the ITT population (analysis set to show superiority), adjudicated stroke or SEE occurred in 296 subjects in the edoxaban 60 mg group (1.57% per year), and 337 subjects in the warfarin group (1.80% per year). Compared to warfarin-treated subjects, the HR in the edoxaban 60 mg group was 0.87 (99% CI: 0.71, 1.07, p = 0.08 for superiority).
In subgroup analyses, for subjects in the 60 mg treatment group who were dose reduced to 30 mg in the ENGAGE AF-TIMI 48 study (for body weight ≤ 60 kg, moderate renal impairment, or concomitant use of P-gp inhibitors), the event rate was: 2.29% per year for the primary endpoint, compared to the event rate of 2.66% per year for the matching subjects in the warfarin group [HR (95% CI): 0.86 (0.66, 1.13)].
The efficacy results for pre-specified major subgroups (with dose reduction as required), including age, body weight, gender, status of renal function, prior stroke or TIA, diabetes and P-gp inhibitors were generally consistent with the primary efficacy results for the overall population studied in the trial.
The Hazard Ratio (Edoxaban 60 mg vs. warfarin) for the primary endpoint in the centres with a lower average time of INR in the target range (INR TTR) for warfarin was 0.73 - 0.80 for the lowest 3 quartiles (INR TTR ≤ 57.7% to ≤ 73.9%). It was 1.07 in centres with the best control of warfarin therapy (4
th quartile with > 73.9% of INR values in the therapeutic range).
There was a statistically significant interaction between the effect of edoxaban versus warfarin on the main study outcome (stroke/SEE) and renal function (p-value 0.0042; mITT, overall study period).
Table 2 shows ischaemic strokes/SEE by creatinine clearance category in NVAF patients in ENGAGE AF-TIMI 48. There is a decreasing event rate at increasing CrCL in both treatment groups. (See Table 2.)
Click on icon to see table/diagram/image
Within renal function subgroups, results for the secondary efficacy endpoints were consistent with those for the primary endpoint.
Superiority testing was performed on the ITT Overall Study Period.
Stroke and SEE occurred in fewer subjects in the edoxaban 60 mg treatment group than in the warfarin group (1.57% and 1.80% per year, respectively), with a HR of 0.87 (99% CI: 0.71, 1.07, p = 0.0807 for superiority).
The pre-specified composite endpoints for the comparison of the edoxaban 60 mg treatment group to warfarin for stroke, SEE, and CV mortality HR (99% CI) was 0.87 (0.76, 0.99), MACE 0.89 (0.78, 1.00), and stroke, SEE, and all-cause mortality 0.90 (0.80, 1.01).
The results for all-cause mortality (adjudicated deaths) in the ENGAGE AF-TIMI 48 study were 769 (3.99% per year) for subjects taking edoxaban 60 mg (30 mg dose reduced) as opposed to 836 (4.35% per year) for warfarin [HR (95% CI): 0.91 (0.83, 1.01)].
All-cause mortality (adjudicated deaths) per renal subgroups (edoxaban vs. warfarin): CrCL 30 to ≤ 50 mL/min [HR (95% CI): 0.81 (0.68, 0.97)]; CrCL > 50 to < 80 mL/min [HR (95% CI): 0.87 (0.75, 1.02)]; CrCL ≥ 80 mL/min [HR (95% CI): 1.15 (0.95, 1.40)].
Edoxaban 60 mg (30 mg dose reduced) resulted in a lower rate of cardiovascular mortality compared to warfarin [HR (95% CI): 0.86 (0.77, 0.97)].
Adjudicated efficacy cardiovascular mortality per renal subgroups (edoxaban vs. warfarin): CrCL 30 to ≤ 50 mL/min [HR (95% CI): 0.80 (0.65, 0.99)]; CrCL > 50 to < 80 mL/min [HR (95% CI): 0.75 (0.62, 0.90)]; CrCL ≥ 80 mL/min [HR (95% CI): 1.16 (0.92, 1.46)].
Safety in patients with NVAF in ENGAGE AF-TIMI 48: The primary safety endpoint was major bleeding.
There was a significant risk reduction in favour of the edoxaban 60 mg treatment group compared with the warfarin group in major bleeding (2.75%, and 3.43% per year, respectively) [HR (95% CI): 0.80 (0.71, 0.91); p = 0.0009], ICH (0.39%, and 0.85% per year, respectively) [HR (95% CI): 0.47 (0.34, 0.63); p < 0.0001], and other types of bleeding (Table 3).
The reduction in fatal bleeds was also significant for the edoxaban 60 mg treatment group compared with the warfarin group (0.21%, and 0.38%) [HR (95% CI): 0.55 (0.36, 0.84); p = 0.0059 for superiority], primarily because of the reduction in fatal ICH bleeds [HR (95% CI): 0.58 (0.35, 0.95); p = 0.0312]. (See Table 3.)
Click on icon to see table/diagram/image
Tables 4, 5 and 6 show major, fatal and intracranial bleedings, respectively, by creatinine clearance category in NVAF patients in ENGAGE AF-TIMI 48. There is a decreasing event rate at increasing CrCL in both treatment group. (See Tables 4, 5, and 6.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In subgroup analyses, for subjects in the 60 mg treatment group who were dose reduced to 30 mg in the ENGAGE AF-TIMI 48 study for body weight ≤ 60 kg, moderate renal impairment, or concomitant use of P-gp inhibitors, 104 (3.05% per year) of edoxaban 30 mg dose reduced subjects and 166 (4.85% per year) of warfarin dose reduced subjects had a major bleeding event [HR (95% CI): 0.63 (0.50, 0.81)].
In the ENGAGE AF-TIMI 48 study there was a significant improvement in Net Clinical Outcome (First Stroke, SEE, Major Bleed, or All-Cause Mortality; mITT population, overall study period) in favour of edoxaban, HR (95% CI): 0.89 (0.83, 0.96); p = 0.0024, when edoxaban 60 mg treatment group was compared to warfarin.
Treatment of DVT, treatment of PE and the prevention of recurrent DVT and PE (VTE): The edoxaban clinical programme for VTE was designed to demonstrate the efficacy and safety of edoxaban in the treatment of DVT and PE, and the prevention of recurrent DVT and PE.
In the pivotal Hokusai-VTE study, 8,292 subjects were randomised to receive initial heparin therapy (enoxaparin or unfractionated heparin) followed by edoxaban 60 mg once daily or the comparator. In the comparator arm, subjects received initial heparin therapy concurrently with warfarin, titrated to a target INR of 2.0 to 3.0, followed by warfarin alone. The treatment duration was from 3 months up to 12 months, determined by the investigator based on the patient's clinical features.
The majority of edoxaban treated patients were Caucasians (69.6%) and Asians (21.0%), 3.8% were Black, 5.3% were categorised as Other race.
The duration of therapy was at least 3 months for 3,718 (91.6%) edoxaban subjects versus 3,727 (91.4%) of warfarin subjects; at least 6 months for 3,495 (86.1%) of edoxaban subjects versus 3,491 (85.6%) of warfarin subjects; and 12 months for 1,643 (40.5%) edoxaban subjects versus 1,659 (40.4%) of warfarin subjects.
The primary efficacy endpoint was the recurrence of symptomatic VTE, defined as the composite of recurrent symptomatic DVT, non-fatal symptomatic PE and fatal PE in subjects during the 12-month study period. Secondary efficacy outcomes included the composite clinical outcome of recurrent VTE and all-cause mortality.
Edoxaban 30 mg once daily was used for subjects with one or more of the following clinical factors: moderate renal impairment (CrCL 30 - 50 mL/min); body weight ≤ 60 kg; concomitant use of specific P-gp inhibitors.
In the Hokusai-VTE study (Table 7) edoxaban was demonstrated to be non-inferior to warfarin for the primary efficacy outcome, recurrent VTE, which occurred in 130 of 4,118 subjects (3.2%) in the edoxaban group versus 146 of 4,122 subjects (3.5%) in the warfarin group [HR (95% CI): 0.89 (0.70, 1.13); p < 0.0001 for non-inferiority]. In the warfarin group, the median TTR (time in therapeutic range, INR 2.0 to 3.0) was 65.6%. For subjects presenting with PE (with or without DVT), 47 (2.8%) of edoxaban and 65 (3.9%) of warfarin subjects had a recurrent VTE [HR (95% CI): 0.73 (0.50, 1.06)]. (See Table 7.)
Click on icon to see table/diagram/image
For the subjects who were dose reduced to 30 mg (predominantly low body weight or renal function) 15 (2.1%) edoxaban and 22 (3.1%) of warfarin subjects had a recurrent VTE [HR (95% CI): 0.69 (0.36, 1.34)].
The secondary composite endpoint of recurrent VTE and all-cause mortality occurred in 138 subjects (3.4%) in the edoxaban group and 158 subjects (3.9%) in the warfarin group [HR (95% CI): 0.87 (0.70, 1.10)].
The results for all-cause mortality (adjudicated deaths) in Hokusai-VTE were 136 (3.3%) for subjects taking edoxaban 60 mg (30 mg dose reduced) as opposed to 130 (3.2%) for warfarin.
In a pre-specified subgroup analysis of PE subjects 447 (30.6%) and 483 (32.2%) of edoxaban and warfarin treated subjects, respectively, were identified as having PE and NT-proBNP ≥ 500 pg/mL. The primary efficacy outcome occurred in 14 (3.1%) and 30 (6.2%) of edoxaban and warfarin subjects, respectively [HR (95% CI): 0.50 (0.26, 0.94)].
The efficacy results for pre-specified major subgroups (with dose reduction as required), including age, body weight, gender and status of renal function were consistent with the primary efficacy results for the overall population studied in the trial.
Safety in patients with VTE (DVT and PE) in Hokusai-VTE: The primary safety endpoint was clinically relevant bleeding (major or clinically relevant non-major).
Table 8 summarises adjudicated bleeding events for the safety analysis set on-treatment period.
There was a significant risk reduction in favour of edoxaban compared with warfarin for the primary safety endpoint of clinically relevant bleeding, a composite of major bleeding or clinically relevant non-major bleeding (CRNM), which occurred in 349 of 4,118 subjects (8.5%) in the edoxaban group and in 423 of 4,122 subjects (10.3%) in the warfarin group [HR (95% CI): 0.81 (0.71, 0.94); p = 0.004 for superiority]. (See Table 8.)
Click on icon to see table/diagram/image
In subgroup analyses, for subjects who were dose reduced to 30 mg in the Hokusai-VTE study for body weight ≤ 60 kg, moderate renal impairment, or concomitant use of P-gp inhibitors, 58 (7.9%) of edoxaban 30 mg dose reduced subjects and 92 (12.8%) of warfarin subjects had a major bleeding or CRNM event [HR (95%): 0.62 (0.44, 0.86)].
In the Hokusai-VTE study the Net Clinical Outcome (Recurrent VTE, Major Bleed, or All-Cause Mortality; mITT population, overall study period) HR (95% CI) was 1.00 (0.85, 1.18) when edoxaban was compared to warfarin.
Patients undergoing cardioversion: A multicentre, prospective, randomised, open-label study with blinded endpoint evaluation (ENSURE-AF) was conducted which randomised 2199 subjects (oral anticoagulant naïve and pre-treated) with non-valvular atrial fibrillation scheduled for cardioversion, to compare edoxaban 60 mg once daily with enoxaparin/warfarin to maintain a therapeutic INR of 2.0 to 3.0 (randomised 1:1), mean TTR on warfarin was 70.8%. A total of 2149 subjects were treated with either edoxaban (N = 1067) or enoxaparin/warfarin (N = 1082). Subjects in the edoxaban treatment group received 30 mg once daily if one or more of the following clinical factors were present: moderate renal impairment (CrCL 30 - 50 mL/min), low body weight (≤ 60 kg) or concomitant use of specific P-gp inhibitors. The majority of subjects in the edoxaban and warfarin groups had cardioversion performed (83.7% and 78.9%, respectively) or were auto-converted (6.6% and 8.6%, respectively). TEE-guided (within 3 days of initiation) or conventional cardioversion (at least 21 days of pre-treatment) was employed. Subjects were maintained on treatment for 28 days post cardioversion.
The primary efficacy outcome consisted of a composite of all stroke, SEE, MI and CV mortality. A total of 5 (0.5%, 95% CI 0.15% - 1.06%) events occurred in subjects in the edoxaban group (N = 1095) and 11 (1.0%, 95% CI 0.50% - 1.78%) events in the warfarin group (N = 1104); OR 0.46 (95% CI 0.12 - 1.43); ITT analysis set overall study period with mean duration of 66 days.
The primary safety outcome was a composite of major and CRNM bleeding. A total of 16 (1.5%, 95% CI 0.86% - 2.42%) events occurred in subjects in the edoxaban (N = 1067) group and 11 (1.0%, 95% CI 0.51% - 1.81%) events in the warfarin (N = 1082) group; OR 1.48 (95% CI 0.64 - 3.55); safety analysis set on-treatment period.
This exploratory study showed low rates of major and CRNM bleeding and thromboembolism in the two treatment groups in the setting of cardioversion.
Pharmacokinetics: Absorption: Edoxaban is absorbed with peak plasma concentrations within 1 - 2 hours. The absolute bioavailability is approximately 62%. Food increases peak exposure to a varying extent, but has minimal effect on total exposure. Edoxaban was administered with or without food in the ENGAGE AF-TIMI 48 and the Hokusai-VTE studies. Edoxaban is poorly soluble at pH of 6.0 or higher. Co-administration of proton-pump inhibitors had no relevant impact on edoxaban exposure.
Distribution: Disposition is biphasic. The volume of distribution is 107 (19.9) L mean (SD).
In vitro plasma protein binding is approximately 55%. There is no clinically relevant accumulation of edoxaban (accumulation ratio 1.14) with once daily dosing. Steady state concentrations are achieved within 3 days.
Biotransformation: Unchanged edoxaban is the predominant form in plasma. Edoxaban is metabolised via hydrolysis (mediated by carboxylesterase 1), conjugation or oxidation by CYP3A4/5 (< 10%). Edoxaban has three active metabolites, the predominant metabolite (M-4), formed by hydrolysis, is active and reaches less than 10% of the exposure of the parent compound in healthy subjects. Exposure to the other metabolites is less than 5%. Edoxaban is a substrate for the efflux transporter P-glycoprotein (P-gp), but not a substrate for uptake transporters such as organic anion transporter polypeptide OATP1B1, organic anion transporters OAT1 or OAT3 or organic cation transporter OCT2. Its active metabolite is a substrate for OATP1B1.
Elimination: In healthy subjects, the total clearance is estimated as 22 (± 3) L/hour; 50% is renally cleared (11 L/hour). Renal clearance accounts for approximately 35% of the administered dose. Metabolism and biliary/intestinal excretion account for the remaining clearance. The t
½ for oral administration is 10 - 14 hours.
Linearity/non-linearity: Edoxaban displays approximately dose-proportional pharmacokinetics for doses of 15 mg to 60 mg in healthy subjects.
Special populations: Elderly: After taking renal function and body weight into account, age had no additional clinically significant effect on edoxaban pharmacokinetics in a population pharmacokinetic analysis of the pivotal Phase 3 study in NVAF (ENGAGE AF-TIMI 48).
Gender: After accounting for body weight, gender had no additional clinically significant effect on edoxaban pharmacokinetics in a population pharmacokinetic analysis of the Phase 3 study in NVAF (ENGAGE AF-TIMI 48).
Ethnic origin: In a population pharmacokinetic analysis of the ENGAGE AF-TIMI 48 study, peak and total exposure in Asian patients and non-Asian patients were comparable.
Renal impairment: The plasma AUCs for subjects with mild (CrCL > 50 - 80 mL/min), moderate (CrCL 30 - 50 mL/min) and severe (CrCL < 30 mL/min but not undergoing dialysis) renal impairment were increased by 32%, 74%, and 72%, respectively, relative to subjects with normal renal function. In patients with renal impairment the metabolite profile changes and a higher quantity of active metabolites are formed. There is a linear correlation between edoxaban plasma concentration and anti-FXa activity regardless of renal function.
Subjects with ESRD undergoing peritoneal dialysis had 93% higher total exposure compared with healthy subjects.
Population PK modeling indicates that exposure approximately doubles in patients with severe renal impairment (CrCL 15 - 29 mL/min) relative to patients with normal renal function.
Anti-FXa activity by CrCL category: Table 9 as follows shows the edoxaban anti-Factor Xa activity by CrCL category for each indication. (See Table 9.)
Click on icon to see table/diagram/image
Although treatment with edoxaban does not require routine monitoring, the effect on anticoagulation can be estimated by a calibrated quantitative anti-Factor Xa assay which may be useful in exceptional situations where knowledge of edoxaban exposure may help to inform clinical decisions, e.g. overdose and emergency surgery (see also Precautions).
Haemodialysis: A 4 hour haemodialysis session reduced total edoxaban exposures by less than 9%.
Hepatic impairment: Patients with mild or moderate hepatic impairment exhibited comparable pharmacokinetics and pharmacodynamics to their matched healthy control group. Edoxaban has not been studied in patients with severe hepatic impairment (see Dosage & Administration).
Body weight: In a population pharmacokinetic analysis of the ENGAGE AF-TIMI 48 study in NVAF, C
max and AUC in patients with median low body weight (55 kg) were increased by 40% and 13%, respectively, as compared with patients with median high body weight (84 kg). In Phase 3 clinical studies (both NVAF and VTE indications) patients with body weight ≤ 60 kg had a 50% edoxaban dose reduction and had similar efficacy and less bleeding when compared to warfarin.
Pharmacokinetic/pharmacodynamic relationship(s): PT, INR, aPTT and Anti-factor Xa correlate linearly with edoxaban concentrations.
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, or phototoxicity.
Reproductive toxicology: Edoxaban showed vaginal haemorrhage at higher doses in rats and rabbits but had no effects in the reproductive performance of parent rats.
In rats, no effects on male or female fertility were seen.
In animal reproduction studies, rabbits showed increased incidence of gallbladder variations at a dosage of 200 mg/kg which is approximately 65 times the maximum recommended human dose (MRHD) of 60 mg/day based on total body surface area in mg/m
2. Increased post-implantation pregnancy losses occurred in rats at 300 mg/kg/day (approximately 49 times the MRHD) and in rabbits at 200 mg/kg/day (approximately 65 times the MRHD) respectively.
Edoxaban was excreted in the breast milk of lactating rats.
Environmental Risk Assessment (ERA): The active substance edoxaban tosilate is persistent in the environment (for instructions on disposal see Special precautions for disposal under Cautions for Usage).




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image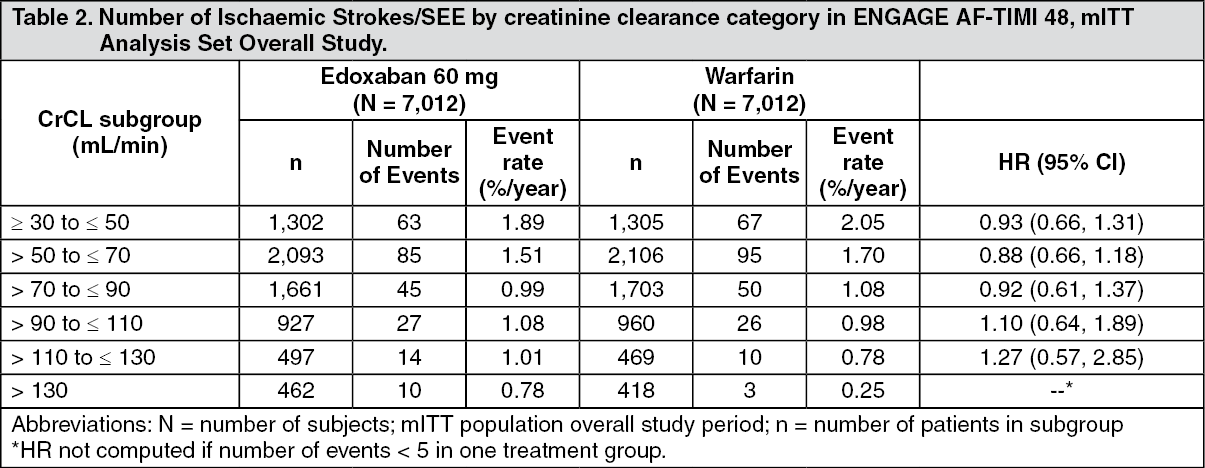 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image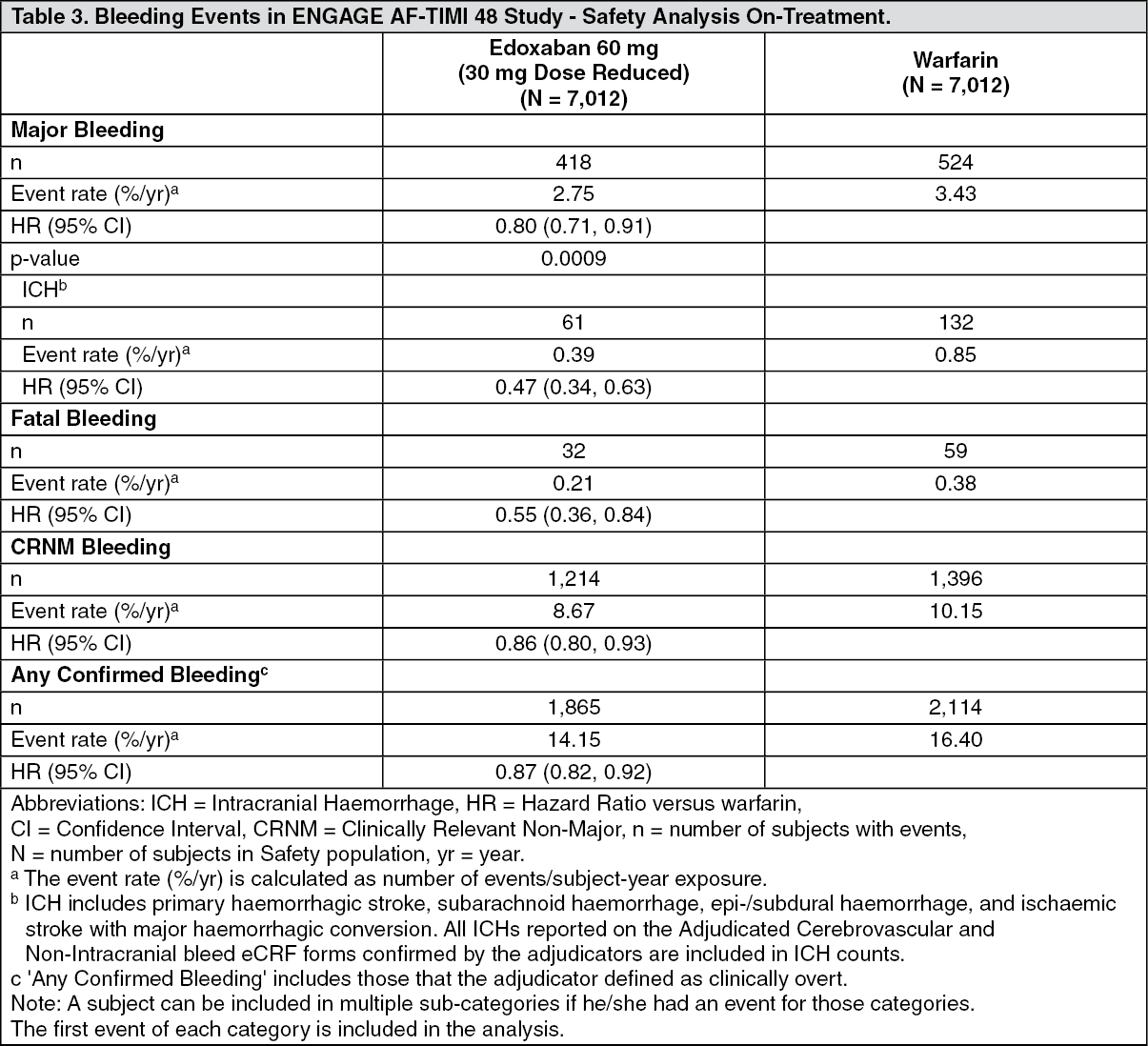 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image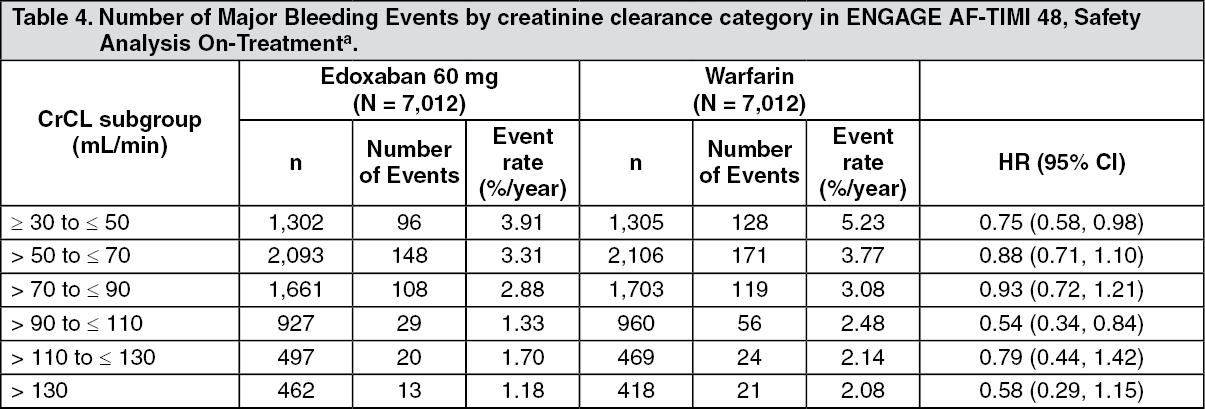 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image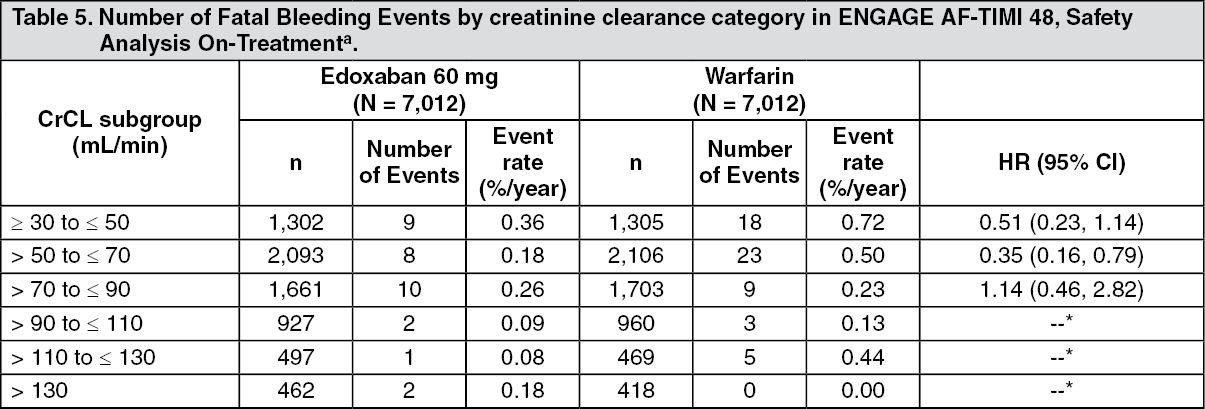 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image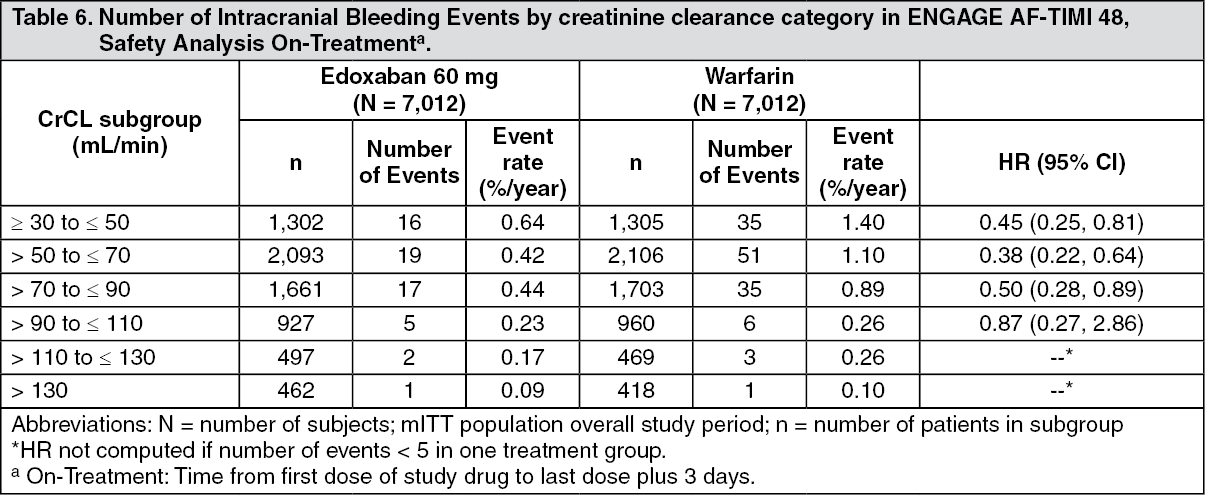 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image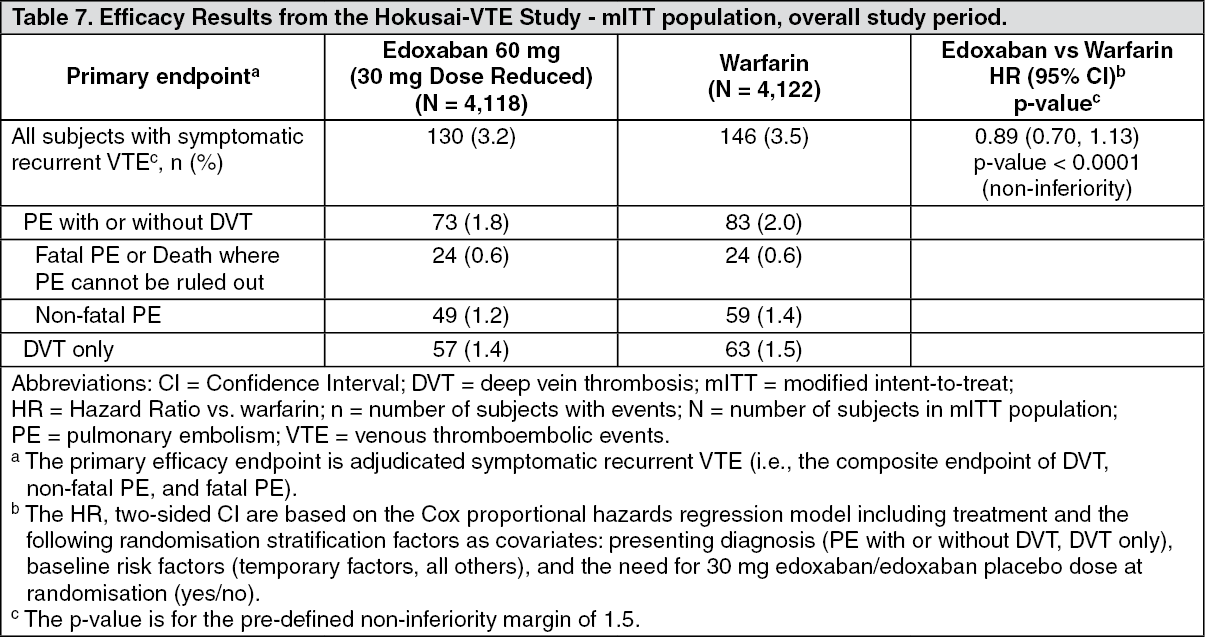 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image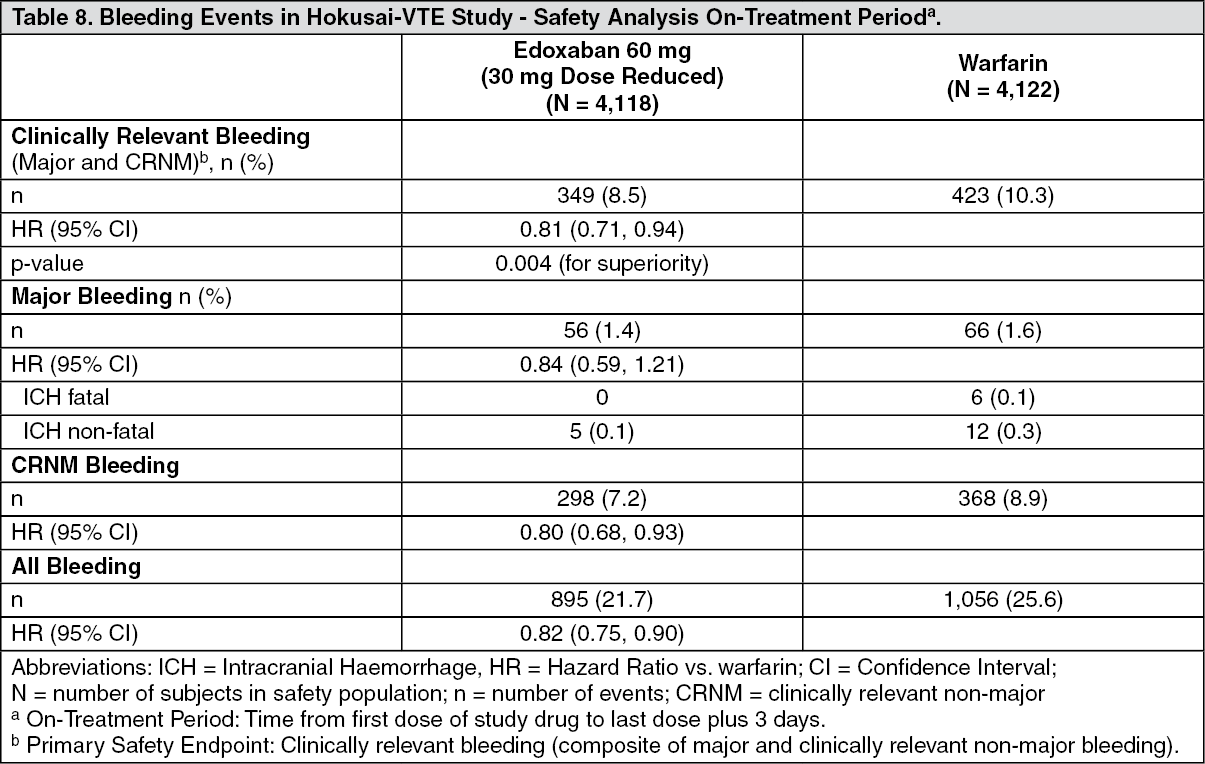 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image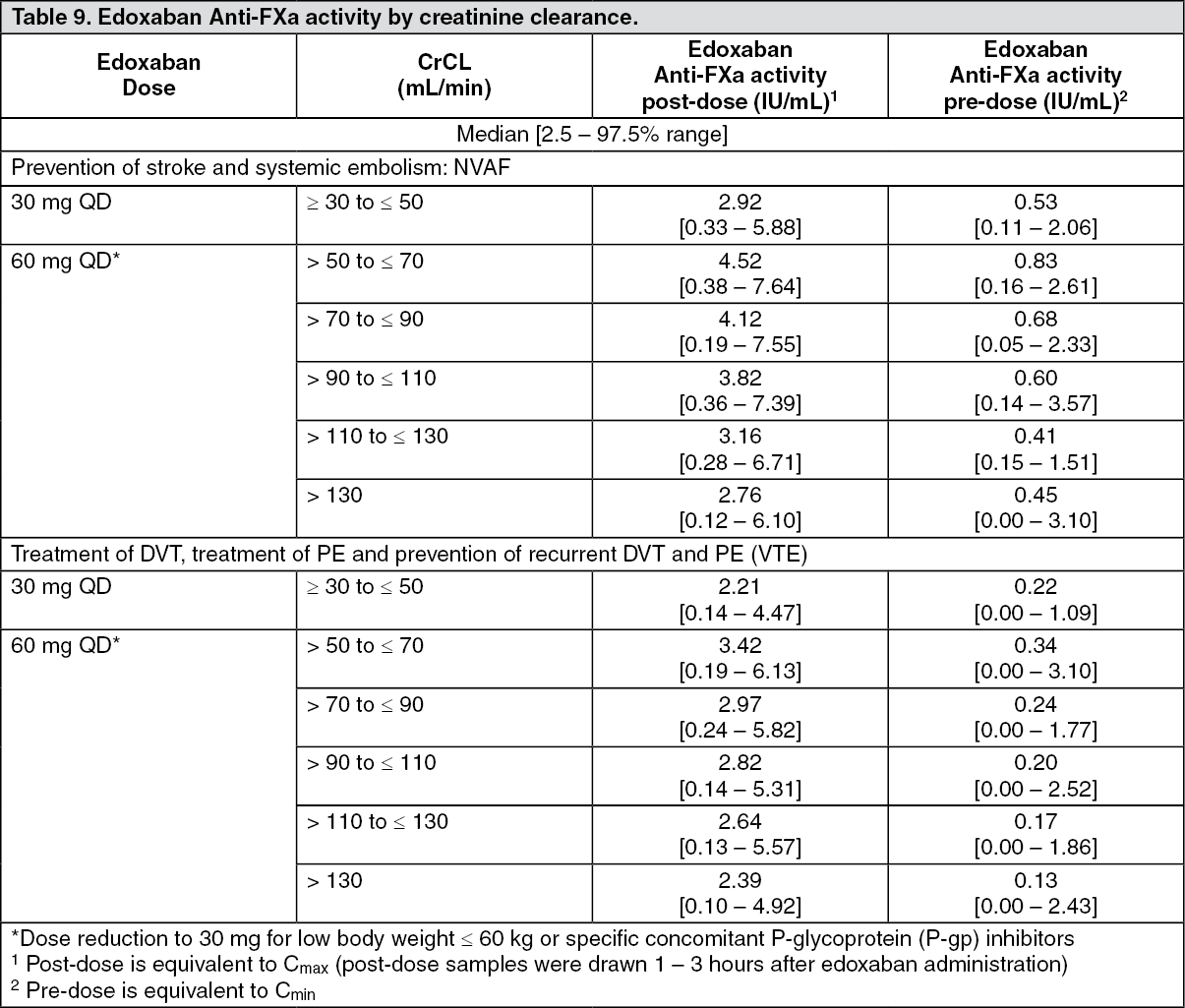 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image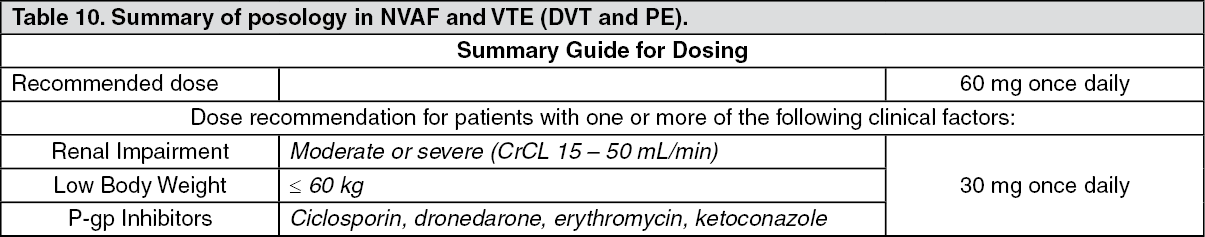 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image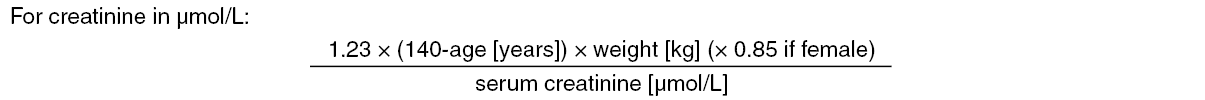 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image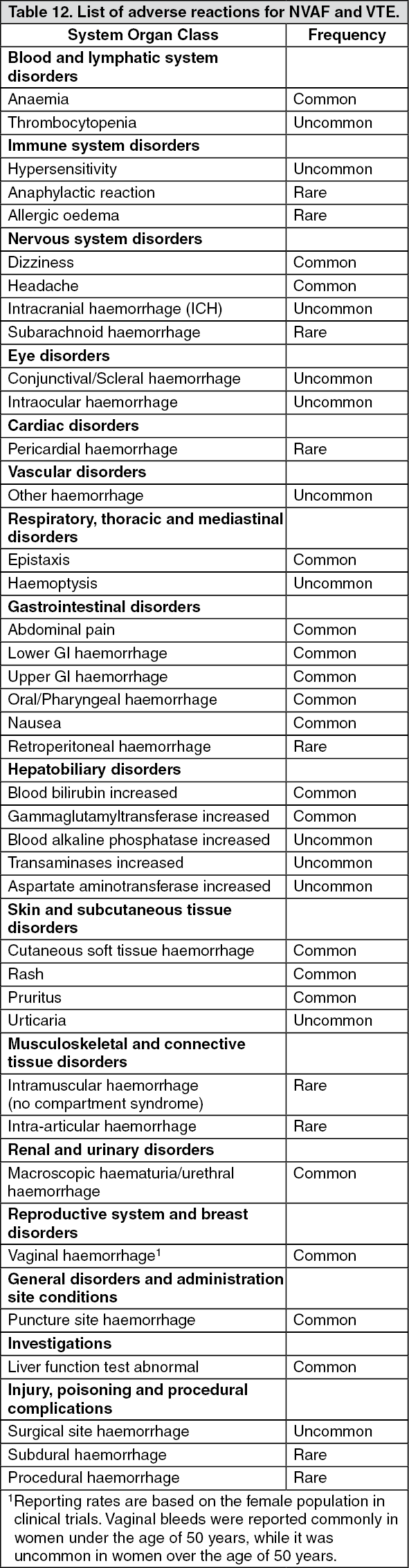 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
