Summary of the safety profile: The safety profile of lenvatinib is based on pooled data from 497 RCC patients treated with lenvatinib in combination with pembrolizumab, including Study 307 (CLEAR), 62 RCC patients treated with lenvatinib in combination with everolimus in Study 205; 458 DTC patients and 496 HCC patients treated with lenvatinib as single-agent therapy.
Lenvatinib in combination with pembrolizumab in RCC: The safety profile of lenvatinib in combination with pembrolizumab is based on data from 497 RCC patients. The most frequently reported adverse reactions (occurring in ≥30% of patients) were diarrhoea (61.8%), hypertension (51.5%) fatigue (47.1%), hypothyroidism (45.1%), decreased appetite (42.1%), nausea (39.6%), stomatitis (36.6%), proteinuria (33.0%), dysphonia (32.8%), and arthralgia (32.4%).
The most common severe (Grade ≥3) adverse reactions (≥5%) were hypertension (26.2%), lipase increased (12.9%), diarrhoea (9.5%), proteinuria (8.0%), amylase increased (7.6%), weight decreased (7.2%), and fatigue (5.2%).
Discontinuation of lenvatinib, pembrolizumab, or both due to an adverse reaction occurred in 33.4% of patients; 23.7% lenvatinib, and 12.9 % both drugs. The most common adverse reactions (≥1%) leading to discontinuation of lenvatinib, pembrolizumab, or both were myocardial infarction (2.4%), diarrhoea (2.0%), proteinuria (1.8%), and rash (1.4%). Adverse reactions that most commonly led to discontinuation of lenvatinib (≥1%) were myocardial infarction (2.2%), proteinuria (1.8%), and diarrhoea (1.0%).
Dose interruptions of lenvatinib, pembrolizumab, or both due to an adverse reaction occurred in 80.1% of patients; lenvatinib was interrupted in 75.3%, and both drugs in 38.6% of patients. Lenvatinib was dose reduced in 68.4% of patients. The most common adverse reactions (≥5%) resulting in dose reduction or interruption of lenvatinib were diarrhoea (25.6%), hypertension (16.1%), proteinuria (13.7%), fatigue (13.1%), appetite decreased (10.9%), palmar-plantar erythrodysaesthesia syndrome (PPE) (10.7%), nausea (9.7%), asthenia (6.6%), stomatitis (6.2%), lipase increased (5.6%), and vomiting (5.6%).
Lenvatinib in combination with everolimus in RCC: The safety profile of lenvatinib in combination with everolimus is based on data from 62 subjects-patients, allowing characterisation only of common adverse drug reactions in RCC patients from Study 205. The adverse reactions presented as follows are based on the combined safety data of 62 RCC patients from Study 205 (see Pharmacology: Pharmacodynamics under Actions) and 458 DTC patients.
The most frequently reported adverse reactions in the DTC and Study 205 RCC patient population (occurring in ≥30% of patients) are diarrhoea (80.6%), hypertension (70.1%)*, fatigue (59.7%), decreased appetite (53.7%), weight decreased (52.6%)*, vomiting (48.4%), nausea (45.2%), proteinuria 38.9%)*, stomatitis (36.9%)*, headache (35.8%)*, dysphonia (35.6%)*, palmar-plantar erythrodysaesthesia syndrome (PPE) (34.1%)* peripheral oedema (33.9%), and hypercholesterolemia (30.6%). Hypertension and proteinuria tend to occur early during lenvatinib treatment (see Description of selected adverse reactions as follows; the asterisked frequencies are from the DTC patient population). In DTC, the majority of Grade 3 to 4 adverse reactions occurred during the first 6 months of treatment except for diarrhoea, which occurred throughout treatment, and weight loss, which tended to be cumulative over time.
The most important serious adverse reactions included renal failure and impairment (11.3%), arterial thromboembolisms (3.9%)*, cardiac failure (1.6%), cerebral haemorrhage (1.6%), intracranial tumour haemorrhage (0.7%)*, PRES/RPLS (0.2%)*, and hepatic failure (0.2%)* (the asterisked frequencies are from the DTC patient population).
In 452 patients with RAI-refractory DTC, dose reduction and discontinuation were the actions taken for an adverse reaction in 63.1% and 19.5% of patients, respectively. Adverse reactions that most commonly led to dose reductions (in ≥5% of patients) were hypertension, proteinuria, diarrhoea, fatigue, PPE, weight decreased, and decreased appetite. Adverse reactions that most commonly led to discontinuation of lenvatinib were proteinuria, asthenia, hypertension, cerebrovascular accident, diarrhoea, and pulmonary embolism.
In RCC Study 205, adverse reactions led to dose reductions in 67.7% of patients and 18 (29.0%) patients discontinued the treatment. The most common adverse reactions (≥5%) resulting in dose reductions in the lenvatinib plus everolimus treated group were diarrhoea (21.0%), thrombocytopenia (6.5%), and vomiting (6.5%).
HCC: The most frequently reported adverse reactions (occurring in ≥30% of patients) are hypertension (44.0%), diarrhoea (38.1%), decreased appetite (34.9%), fatigue (30.6%), and decreased weight (30.4%).
The most important serious adverse reactions were hepatic failure (2.8%), hepatic encephalopathy (4.6%), oesophageal varices haemorrhage (1.4%), cerebral haemorrhage (0.6%), arterial thromboembolic events (2.0%) including myocardial infarction (0.8%), cerebral infarction (0.4%) and cerebrovascular accident (0.4%) and renal failure/impairment events (1.4%). There was a higher incidence of decreased neutrophil count in patients with HCC (8.7% on lenvatinib than in other non-HCC tumour types (1.4%)), which was not associated with infection, sepsis or bacterial peritonitis.
In 496 patients with HCC, dose modification (interruption or reduction) and discontinuation were the actions taken for an adverse reaction in 62.3% and 20.2% of patients, respectively. Adverse reactions that most commonly led to dose modifications (in ≥5% of patients) were decreased appetite, diarrhoea, proteinuria, hypertension, fatigue, PPE and decreased platelet count. Adverse reactions that most commonly led to discontinuation of lenvatinib were hepatic encephalopathy, fatigue, increased blood bilirubin, proteinuria and hepatic failure.
EC: The safety of lenvatinib in combination with pembrolizumab has been evaluated in 530 patients with advanced EC receiving 20 mg lenvatinib once daily and 200 mg pembrolizumab every 3 weeks. The most common (occurring in ≥20% of patients) adverse reactions were hypertension (63%), diarrhoea (57%), hypothyroidism (56%), nausea (51%), decreased appetite (47%), vomiting (39%), fatigue (38%), decreased weight (35%), arthralgia (33%), proteinuria (29%), constipation (27%), headache (27%), urinary tract infection (27%), dysphonia (25%), abdominal pain (23%), asthenia (23%), palmar-plantar erythrodysaesthesia syndrome (23%), stomatitis (23%), anaemia (22%), and hypomagnesaemia (20%).
The most common (occurring in ≥5% of patients) severe (Grade ≥3) adverse reactions were hypertension (37.2%), decreased weight (9.1%), diarrhoea (8.1%), increased lipase (7.7%), decreased appetite (6.4%), asthenia (6%), fatigue (6%), hypokalaemia (5.7%), anaemia (5.3%), and proteinuria (5.1%).
Discontinuation of lenvatinib occurred in 30.6% of patients, and discontinuation of both lenvatinib and pembrolizumab occurred in 15.3% of patients due to an adverse reaction. The most common (occurring in ≥1% of patients) adverse reactions leading to discontinuation of lenvatinib were hypertension (1.9%), diarrhoea (1.3%), asthenia (1.3%), decreased appetite (1.3%), proteinuria (1.3%), and decreased weight (1.1%).
Dose interruption of lenvatinib due to an adverse reaction occurred in 63.2% of patients. Dose interruption of lenvatinib and pembrolizumab due to an adverse reaction occurred in 34.3% of patients. The most common (occurring in ≥5% of patients) adverse reactions leading to interruption of lenvatinib were hypertension
(12.6%), diarrhoea (11.5%), proteinuria (7.2%), vomiting (7%), fatigue (5.7%), and decreased appetite (5.7%).
Dose reduction of lenvatinib due to adverse reactions occurred in 67.0% of patients. The most common (occurring in ≥5% of patients) adverse reactions resulting in dose reduction of lenvatinib were hypertension (16.2%), diarrhoea (12.5%), palmar-plantar erythrodysaesthesia syndrome (9.1%), fatigue (8.7%), proteinuria (7.7%), decreased appetite (6.6%), nausea (5.5%), asthenia (5.1%), and decreased weight (5.1%).
Tabulated list of adverse reactions for DTC, RCC, HCC and EC studies: Similar adverse reactions were observed in clinical trials in DTC, RCC and HCC. Adverse reactions that occur more frequently with lenvatinib and everolimus combination therapy compared to lenvatinib monotherapy are hypothyroidism, (including increased blood thyroid stimulating hormone), hypercholesterolaemia, and severe diarrhoea.
Adverse reactions that occured more frequently with lenvatinib and pembrolizumab combination therapy compared to lenvatinib monotherapy were hypothyroidism (including increased blood thyroid stimulating hormone), hypercholesterolaemia, diarrhoea, lipase increased, amylase increased, rash (including maculopapular rash), and blood creatinine increased.
The safety profile of lenvatinib as combination therapy is based on data from 530 EC patients treated with lenvatinib in combination with pembrolizumab (see Pharmacology: Pharmacodynamics under Actions).
Adverse reactions observed in clinical trials and reported from post-marketing use of lenvatinib are listed in Table 12. The adverse reaction frequency category represents the most conservative estimate of frequency from the individual populations. Adverse reactions known to occur with lenvatinib or combination therapy components given alone may occur during treatment with these medicinal products in combination, even if these reactions were not reported in clinical studies with combination therapy.
For additional safety information when lenvatinib is administered in combination, refer to the package insert for the respective combination therapy components.
Frequencies are defined as: Very common (≥1/10), Common (≥1/100 to <1/10), Uncommon (≥1/1,000 to <1/100), Rare (≥1/10,000 to <1/1,000), Very rare (<1/10,000), Not known (cannot be estimated from the available data).
Within each frequency category, undesirable effects are presented in order of decreasing seriousness. (See Tables 12a and 12b.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Description of selected adverse reactions: Hypertension (see Precautions):
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Description of selected adverse reactions: Hypertension (see Precautions): In CLEAR (see Pharmacology: Pharmacodynamics under Actions), hypertension was reported in 56.3% of patients in the lenvatinib plus pembrolizumab-treated group and 42.6% of patients in the sunitinib-treated group. The exposure-adjusted frequency of hypertension was 0.65 episodes per patient year in the lenvatinib plus pembrolizumab-treated group and 0.73 episodes per patient year in the sunitinib-treated group. The median time to onset in lenvatinib plus pembrolizumab-treated patients was 0.7 months. Reactions of Grade 3 or higher occurred in 28.7% of lenvatinib plus pembrolizumab-treated group compared with 19.4% of the sunitinib-treated group. 16.8% of patients with hypertension had dose modifications of lenvatinib (9.1% dose interruption and 11.9% dose reduction). In 0.9% of patients, hypertension led to permanent treatment discontinuation of lenvatinib.
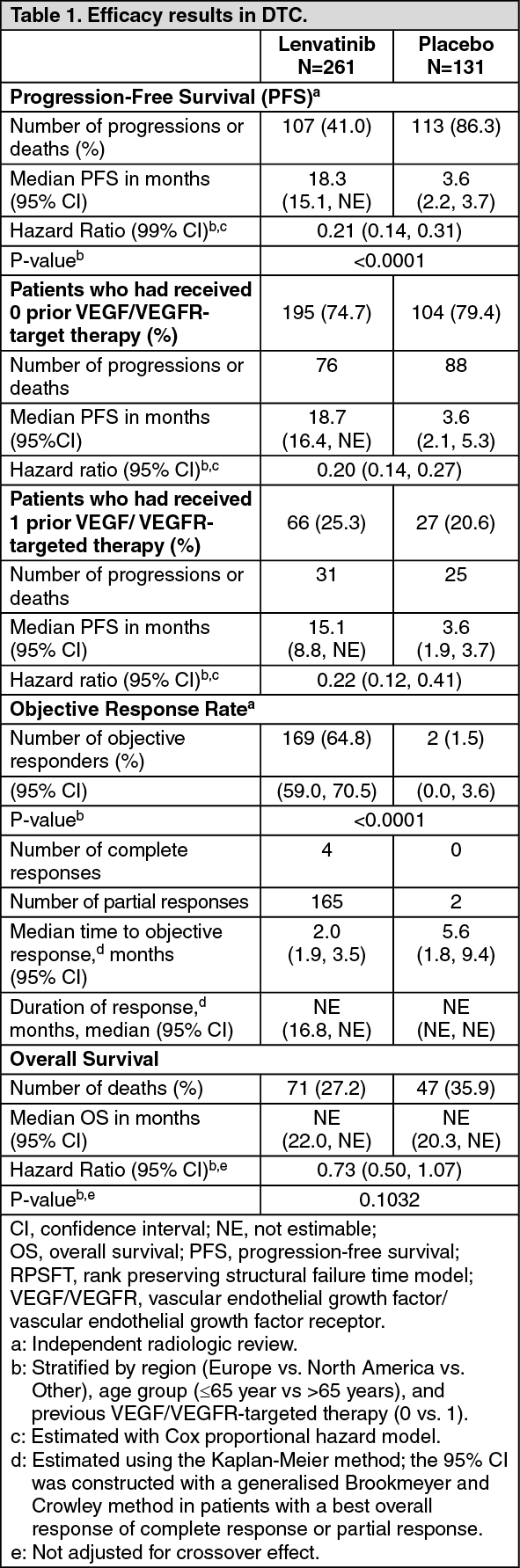
In DTC Study 303 (see Pharmacology: Pharmacodynamics under Actions), hypertension (including hypertension, hypertensive crisis, blood pressure diastolic increased, and blood pressure increased) was reported in 72.8% of lenvatinib-treated patients and 16.0% of patients in the placebo-treated group. The median time to onset in lenvatinib-treated patients was 16 days. Reactions of Grade 3 or higher (including 1 reaction of Grade 4) occurred in 44.4% of lenvatinib-treated patients compared with 3.8% of placebo-treated patients. The majority of cases recovered or resolved following dose interruption or reduction, which occurred in 13.0% and 13.4% of patients, respectively. In 1.1% of patients, hypertension led to permanent treatment discontinuation.
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), hypertension was reported in 41.9% of patients in the lenvatinib plus everolimus-treated group (the incidence of Grade 3 or Grade 4 hypertension was 12.9%) and 10.0% of patients in the everolimus-treated group (the incidence of Grade 3 or Grade 4 hypertension was 2.0%). The median time to onset was 4.9 weeks (any grade) and 6.9 weeks (Grade ≥3) in the lenvatinib plus everolimus-treated group.
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), hypertension (including hypertension, increased blood pressure, increased diastolic blood pressure and orthostatic hypertension) was reported in 44.5% of lenvatinib-treated patients and Grade 3 hypertension occurred in 23.5%. The median time to onset was 26 days. The majority of cases recovered following dose interruption or reduction, which occurred in 3.6% and 3.4% of patients respectively. One subject (0.2%) discontinued lenvatinib due to hypertension.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), hypertension was reported in 65% of patients in the lenvatinib plus pembrolizumab group. Reactions of Grade 3 or higher occurred in 38.4% of patients in the lenvatinib plus pembrolizumab group. The median time to onset in the lenvatinib plus pembrolizumab group was 15 days. Dose interruption, reduction and discontinuation of lenvatinib occurred in 11.6%, 17.7% and 2.0% of patients, respectively.
Proteinuria (see Precautions): In the DTC study (see Pharmacology: Pharmacodynamics under Actions), proteinuria was reported in 33.7% of lenvatinib-treated patients and 3.1% of patients in the placebo-treated group. The median time to onset was 6.7 weeks. Grade 3 reactions occurred in 10.7% of lenvatinib-treated patients and none in placebo-treated patients. The majority of cases had an outcome of recovered or resolved following dose interruption or reduction, which occurred in 16.9% and 10.7% of patients, respectively. Proteinuria led to permanent treatment discontinuation in 0.8% of patients.
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), proteinuria was reported in 30.6% of patients in the lenvatinib plus everolimus-treated group (8.1% were Grade ≥3) and 14.0% of patients in the everolimus-treated group (2.0% were Grade ≥3). The median time to onset of proteinuria was 6.1 weeks (any grade) and 20.1 weeks (Grade ≥3) in the lenvatinib plus everolimus-treated group. Proteinuria led to permanent treatment discontinuation in 4.8% of patients.
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), proteinuria was reported in 26.3% of lenvatinib-treated patients and Grade 3 reactions occurred in 5.9%. The median time to onset was 6.1 weeks. The majority of cases recovered following dose interruption or reduction, which occurred in 6.9% and 2.5% of patients respectively. Proteinuria led to permanent treatment discontinuation in 0.6% of patients.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), proteinuria was reported in 29.6% of lenvatinib plus pembrolizumab-treated patients and Grade ≥3 reactions occurred in 5.4% of patients. The median time to onset was 34.5 days. Dose interruption, reduction and discontinuation of lenvatinib occurred in 6.2%, 7.9% and 1.2% of patients, respectively.
Renal failure and impairment (see Precautions): In the DTC study (see Pharmacology: Pharmacodynamics under Actions), 5.0% of patients developed renal failure and 1.9% developed renal impairment (3.1% of patients had a Grade ≥3 event of renal failure or impairment). In the placebo group 0.8% of patients developed renal failure or impairment (0.8% were Grade ≥3).
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), 8.1% of patients in the lenvatinib plus everolimus treated group developed renal failure and 3.2% developed renal impairment, (9.7% of patients had a Grade 3 event of renal failure or impairment). In the everolimus monotherapy group 2.0% of patients developed renal failure (2.0% were Grade 3).
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), 7.1% of lenvatinib-treated patients developed a renal failure/impairment event. Grade 3 or greater reactions occurred in 1.9% of lenvatinib-treated patients.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), 18.2% of lenvatinib plus pembrolizumab-treated patients developed a renal failure/impairment event. Grade ≥3 reactions occurred in 4.2% of patients. The median time to onset
was 86.0 days. Dose interruption, reduction and discontinuation of lenvatinib occurred in 3.0%, 1.7% and 1.2% of patients, respectively.
Cardiac dysfunction (see Precautions): In the DTC study (see Pharmacology: Pharmacodynamics under Actions), decreased ejection fraction/cardiac failure was reported in 6.5% of patients (1.5% were Grade ≥3) in the lenvatinib treated group, and 2.3% in the placebo group (none were Grade ≥3).
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), decreased ejection fraction/cardiac failure was reported in 4.8% of patients (3.2% were Grade ≥3) in the lenvatinib plus everolimus treated group, and 4.0% in the everolimus group (2.0% were Grade ≥3). The median time to onset of decreased ejection fraction and cardiac failure was 15.7 weeks (any grade) and 32.8 weeks (Grade ≥3) in the lenvatinib plus everolimus-treated group.
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), cardiac dysfunction (including congestive cardiac failure, cardiogenic shock, and cardiopulmonary failure) was reported in 0.6% of patients (0.4% were Grade ≥3) in the lenvatinib-treated group.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), cardiac dysfunction was reported in 1.0% of lenvatinib plus pembrolizumab-treated patients and Grade ≥3 reactions occurred in 0.5% of patients. The median time to onset
was 112.0 days. Dose reduction and discontinuation of lenvatinib both occurred in 0.2% of patients.
Posterior reversible encephalopathy syndrome (PRES)/Reversible posterior leucoencephalopathy syndrome (RPLS) (see Precautions): In the DTC study (see Pharmacology: Pharmacodynamics under Actions), there was 1 event of PRES (Grade 2) in the lenvatinib-treated group and no reports in the placebo group.
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), there was 1 event of PRES (Grade 3) in the lenvatinib-treated group, occurring after 18.4 weeks of treatment. There were no reports in the lenvatinib plus everolimus or everolimus monotherapy groups.
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), there was 1 event of PRES (Grade 2) in the lenvatinib-treated group.
Amongst 1,823 patients treated with lenvatinib monotherapy in clinical trials, there were 5 cases (0.3%) of PRES (0.2% were Grade 3 or 4), all of which resolved after treatment and/or dose interruption, or permanent discontinuation.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), there was one event of PRES (Grade 1) in the lenvatinib plus pembrolizumab-treated group for which lenvatinib was interrupted.
Hepatotoxicity (see Precautions): In CLEAR (see Pharmacology: Pharmacodynamics under Actions), the most commonly reported liver related adverse reactions in the lenvatinib plus pembrolizumab treated group were elevations of liver enzyme levels, including increases in alanine aminotransferase (11.9%), aspartate aminotransferase (11.1%) and blood bilirubin (4.0%). Similar events occurred in the sunitinib-treated group at rates of 10.3%, 10.9% and 4.4% respectively. The median time to onset of liver events was 3.0 months (any grade) in the lenvatinib plus pembrolizumab treated group and 0.7 months in the sunitinib-treated group. The exposure-adjusted frequency of hepatoxicity events was 0.39 episodes per patient year in the lenvatinib plus pembrolizumab-treated group and 0.46 episodes per patient year in the sunitinib-treated group. Grade 3 liver related reactions occurred in 9.9% of lenvatinib plus pembrolizumab treated patients and 5.3% of sunitinib-treated patients. Liver related reactions led to dose interruptions and reductions of lenvatinib in 8.5% and 4.3% of patients, respectively, and to permanent discontinuation of lenvatinib in 1.1% of patients.
In the DTC study (see Pharmacology: Pharmacodynamics under Actions), the most commonly reported liver-related adverse reactions were hypoalbuminaemia (9.6% lenvatinib vs. 1.5% placebo) and elevations of liver enzyme levels, including increases in alanine aminotransferase (7.7% lenvatinib vs. 0 placebo), aspartate aminotransferase (6.9% lenvatinib vs. 1.5% placebo), and blood bilirubin (1.9% lenvatinib vs. 0 placebo). The median time to onset of liver reactions in lenvatinib-treated patients was 12.1 weeks. Liver-related reactions of Grade 3 or higher (including 1 Grade 5 case of hepatic failure) occurred in 5.4% of lenvatinib-treated patients compared with 0.8% in placebo-treated patients. Liver-related reactions led to dose interruptions and reductions in 4.6% and 2.7% of patients, respectively, and to permanent discontinuation in 0.4%.
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), the most commonly reported liver-related adverse reactions in the lenvatinib plus everolimus-treated group were elevations of liver enzyme levels, including increases in alanine aminotransferase (9.7%), aspartate aminotransferase (4.8%), alkaline phosphatase (4.8%), and blood bilirubin (3.2%). The median time to onset of liver events was 6.7 weeks (any grade) and 14.2 weeks (Grade ≥3) in the lenvatinib plus everolimus-treated group. Grade 3 liver-related reactions occurred in 3.2% of lenvatinib plus everolimus-treated patients. Liver-related reactions led to dose interruptions and reductions in 1.6% and 1.6% of patients, respectively, and to permanent discontinuation in 3.2% of patients.
Amongst 1,166 patients treated with lenvatinib, there were 3 cases (0.3%) of hepatic failure, all with a fatal outcome. One occurred in a patient with no liver metastases. There was also a case of acute hepatitis in a patient without liver metastases.
In the Phase 3 REFLECT trial (see Pharmacology: Pharmacodynamics under Actions), the most commonly reported hepatotoxicity adverse reactions were increased blood bilirubin (14.9%), increased aspartate aminotransferase (13.7%), increased alanine aminotransferase (11.1%), hypoalbuminaemia (9.2%), hepatic encephalopathy (8.0%), increased gamma-glutamyltransferase (7.8%) and increased blood alkaline phosphatase (6.7%). The median time to onset of hepatotoxocity adverse reactions was 6.4 weeks. Hepatotoxicity reactions of ≥Grade 3 occurred in 26.1% of lenvatinib-treated patients. Hepatic failure (including fatal events in 12 patients) occurred in 3.6% of patients (all were ≥Grade 3). Hepatic encephalopathy (including fatal events in 4 patients) occurred in 8.4% of patients (5.5% were ≥Grade 3). There were 17 (3.6%) deaths due to hepatotoxicity events in the lenvatinib arm and 4 (0.8%) deaths in the sorafenib arm. Hepatotoxicity adverse reactions led to dose interruptions and reductions in 12.2% and 7.4% of lenvatinib-treated patients respectively, and to permanent discontinuation in 5.5%.
Across clinical studies in which 1327 patients received lenvatinib monotherapy in indications other than HCC, hepatic failure (including fatal events) was reported in 4 patients (0.3%), liver injury in 2 patients (0.2%), acute hepatitis in 2 patients (0.2%), and hepatocellular injury in 1 patient (0.1%).
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), hepatotoxicity was reported in 33.7% of lenvatinib plus pembrolizumab-treated patients and Grade ≥3 reactions occurred in 12.1% of patients. The median time to onset was 56.0 days. Dose interruption, reduction and discontinuation of lenvatinib occurred in 5.2%, 3.0% and 1.2% of patients, respectively.
Arterial thromboembolisms (see Precautions): In CLEAR (see Pharmacology: Pharmacodynamics under Actions), 5.4% of patients in the lenvatinib plus pembrolizumab treated group reported arterial thromboembolic events (of which 3.7% were Grade ≥3) compared with 2.1% of patients in the sunitinib-treated group (of which 0.6% were Grade ≥3). No events were fatal. The exposure-adjusted frequency of arterial thromboembolic event episodes was 0.04 episodes per patient year in the lenvatinib plus pembrolizumab-treated group and 0.02 episodes per patient year in the sunitinib-treated group. The most commonly reported arterial thromboembolic event in the lenvatinib plus pembrolizumab treated group was myocardial infarction (3.4%). One event of myocardial infarction (0.3%) occurred in the sunitinib-treated group. The median time to onset of arterial thromboembolic events was 10.4 months in the lenvatinib plus pembrolizumab treated group.
In the DTC study (see Pharmacology: Pharmacodynamics under Actions), arterial thromboembolic events were reported in 5.4% of lenvatinib-treated patients and 2.3% of patients in the placebo group.
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), 1.6% of patients in the lenvatinib plus everolimus-treated group reported arterial thromboembolic events. The time to onset was 69.6 weeks. In the everolimus group, 6.0% of patients reported an arterial thromboembolism (4.0% were Grade ≥3).
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), arterial thromboembolic events reported in 2.3% of patients treated with lenvatinib.
Amongst 1,823 patients treated with lenvatinib monotherapy in clinical studies, there were 10 cases (0.5%) of arterial thromboembolisms (5 cases of myocardial infarction and 5 cases of cerebrovascular accident) with a fatal outcome.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), arterial thromboembolisms were reported in 3.7% of lenvatinib plus pembrolizumab-treated patients and Grade ≥3 reactions occurred in 2.2% of patients. The median time to onset was 59.0 days. Dose interruption and discontinuation of lenvatinib occurred in 0.2% and 2.0% of patients, respectively.
Haemorrhage (see Precautions): In the DTC study (see Pharmacology: Pharmacodynamics under Actions), haemorrhage was reported in 34.9% (1.9% were Grade ≥3) of lenvatinib-treated patients versus 18.3% (3.1% were Grade ≥3) of placebo-treated patients. Reactions that occurred at an incidence of ≥0.75% above placebo were: epistaxis (11.9%), haematuria (6.5%), contusion (4.6%), gingival bleeding (2.3%), haematochezia (2.3%), rectal haemorrhage (1.5%), haematoma (1.1%), haemorrhoidal haemorrhage (1.1%), laryngeal haemorrhage (1.1%), petechiae (1.1%), and intracranial tumour haemorrhage (0.8%). In this trial, there was 1 case of fatal intracranial haemorrhage among 16 patients who received lenvatinib and had CNS metastases at baseline.
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), haemorrhage was reported in 38.7% (8.1% were Grade ≥3) of patients in the lenvatinib plus everolimus-treated group. Reactions that occurred at an incidence of ≥2.0% were: epistaxis (22.6%), haematuria (4.8%), haematoma (3.2%), and gastric haemorrhage (3.2%). The median time to first onset of was 10.2 weeks (any grade) and 7.6 weeks (Grade ≥3) in the lenvatinib plus everolimus-treated group. The incidence of serious haemorrhage was 4.8% (cerebral haemorrhage, gastric haemorrhage and haemarthrosis). Discontinuation due to haemorrhagic events occurred in 3.2% of patients in the lenvatinib plus everolimus-treated group. There was one case of fatal cerebral haemorrhage in the lenvatinib plus everolimus-treated group and one case of fatal intracranial haemorrhage in the lenvatinib-treated group.
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), haemorrhage was reported in 24.6% of patients and 5.0% were Grade ≥3. Grade 3 reactions occurred in 3.4%, Grade 4 reactions in 0.2% and 7 patients (1.5%) had a grade 5 reaction including cerebral haemorrhage, upper gastrointestinal haemorrhage, intestinal haemorrhage and tumour haemorrhage. The median time to first onset was 11.9 weeks. A haemorrhage event led to dose interruption or reduction in 3.2% and 0.8% patients respectively and to treatment discontinuation in 1.7% of patients.
Across clinical studies in which 1,327 patients received lenvatinib monotherapy in indications other than HCC, Grade ≥3 or greater haemorrhage was reported in 2% of patients, 3 patients (0.2%) had a Grade 4 haemorrhage and 8 patients (0.6%) had a Grade 5 reaction including arterial haemorrhage, haemorrhagic stroke, intracranial haemorrhage, intracranial tumour haemorrhage, haematemesis, melaena, haemoptysis and tumour haemorrhage.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), haemorrhage was reported in 24.4% of lenvatinib plus pembrolizumab-treated patients and Grade ≥3 reactions occurred in 3.0% of patients. The median time to onset was 65.0 days. Dose interruption, reduction and discontinuation of lenvatinib occurred in 1.7%, 1.2% and 1.7% of patients, respectively.
Hypocalcaemia (see QT interval prolongation under Precautions): In the DTC study (see Pharmacology: Pharmacodynamics under Actions), hypocalcaemia was reported in 12.6% of lenvatinib-treated patients vs. no cases in the placebo arm. The median time to first onset in lenvatinib-treated patients was 11.1 weeks. Reactions of Grade 3 or 4 severity occurred in 5.0% of lenvatinib-treated vs 0 placebo-treated patients. Most reactions resolved following supportive treatment, without dose interruption or reduction, which occurred in 1.5% and 1.1% of patients, respectively; 1 patient with Grade 4 hypocalcaemia discontinued treatment permanently.
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), hypocalcaemia was reported in 8.1% of patients in the lenvatinib plus everolimus-treated group (3.2% were Grade ≥3) and 4.0% of patients in the everolimus-treated group (none were Grade ≥3). The median time to onset of hypocalcaemia was 28.3 weeks (any grade) and 45.9 weeks (Grade ≥3) in the lenvatinib plus everolimus-treated group. There was one Grade 4 TEAE. No events of hypocalcaemia required dose reduction or interruption, and no patients discontinued treatment due to hypocalcaemia.
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), hypocalcaemia was reported in 1.1% of patients, with grade 3 reactions occurring in 0.4%. Lenvatinib dose interruption due to hypocalcaemia occurred in one subject (0.2%) and there were no dose reductions or discontinuations.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), hypocalcaemia was reported in 3.9% of lenvatinib plus pembrolizumab-treated patients and Grade ≥3 reactions occurred in 1.0% of patients. The median time to onset was 148.0 days. No lenvatinib dose modifications were reported.
Gastrointestinal perforation and fistula formation (see Precautions): In the DTC study (see Pharmacology: Pharmacodynamics under Actions), events of gastrointestinal perforation or fistula were reported in 1.9% of lenvatinib-treated patients and 0.8% of patients in the placebo group.
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), 1.6% of cases of perforated appendicitis (of Grade 3) occurred in the lenvatinib plus everolimus-treated group; there were no reports in the lenvatinib or everolimus groups.
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), events of gastrointestinal perforation or fistula were reported in 1.9% of lenvatinib-treated patients.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), events of fistula formation were reported in 2.5% of lenvatinib plus pembrolizumab-treated patients and Grade ≥3 reactions occurred in 2.5% of patients. The median time to onset was 117.0 days. Discontinuation of lenvatinib occurred in 1.0% of patients. Events of gastrointestinal perforation were reported in 3.9% of lenvatinib plus pembrolizumab-treated patients and Grade ≥3 reactions occurred in 3.0% of patients. The median time to onset was 42 days. Dose interruption and discontinuation of lenvatinib occurred in 0.5% and 3.0% of patients, respectively.
Non-Gastrointestinal fistula (see Precautions): Lenvatinib use has been associated with cases of fistula including reactions resulting in death. Reports of fistula that involve areas of the body other than stomach or intestines were observed across various indications. Reactions were reported at various time points during treatment ranging from two weeks to greater than 1 year from initiation of lenvatinib, with median latency of about 3 months.
QT interval prolongation (see Precautions): In the DTC study (see Pharmacology: Pharmacodynamics under Actions), QT/QTc interval prolongation was reported in 8.8% of lenvatinib-treated patients and 1.5% of patients in the placebo group. The incidence of QT interval prolongation of greater than 500 ms was 2% in the lenvatinib-treated patients compared to no reports in the placebo group.
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), QTc interval increases greater than 60 ms were reported in 11% of patients in the lenvatinib plus everolimus-treated group. The incidence of QTc interval greater than 500 ms was 6% in the lenvatinib plus everolimus-treated group. No reports of QTc interval prolongation greater than 500 ms or increases greater than 60 ms occurred in the everolimus-treated group.
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), QT/QTc interval prolongation was reported in 6.9% of lenvatinib-treated patients. The incidence of QTcF interval prolongation of greater than 500 ms was 2.4%.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), QT interval prolongation was reported in 3.9% of lenvatinib plus pembrolizumab-treated patients and Grade ≥3 reactions occurred in 0.5% of patients.
The median time to onset was 115.5 days. Dose interruption and reduction of lenvatinib occurred in 0.2% and 0.5% of patients, respectively.
Increased Blood thyroid stimulating hormone (see Precautions): In CLEAR (see Pharmacology: Pharmacodynamics under Actions), hypothyroidism occurred in 47.2% of patients in the lenvatinib plus pembrolizumab treated group and 26.5% of patients in the sunitinib treated group. The exposure-adjusted frequency of hypothyroidism was 0.39 episodes per patient year in the lenvatinib plus pembrolizumab-treated group and 0.33 episodes per patient year in the sunitinib-treated group. In general, the majority of hypothyroidism events in the lenvatinib plus pembrolizumab treated group were of Grade 1 or 2. Grade 3 hypothyroidism was reported in 1.4% of patients in the lenvatinib plus pembrolizumab treated group versus none in the sunitinib-treated group. At baseline, 90% of patients in the lenvatinib plus pembrolizumab-treated group and 93.1% of patients in the sunitinib-treated group had baseline TSH levels ≤ upper limit of normal.
Elevations of TSH > upper limit of normal were observed post baseline in 85.0% of lenvatinib plus pembrolizumab treated patients versus 65.6% of sunitinib-treated patients. In lenvatinib plus pembrolizumab-treated patients, hypothyroidism events resulted in dose modification of lenvatinib (reduction or interruption) in 2.6% patients and discontinuation of lenvatinib in 1 patient.
In the DTC study (see Pharmacology: Pharmacodynamics under Actions), 88% of all patients had a baseline TSH level less than or equal to 0.5 mU/L. In those patients with a normal TSH at baseline, elevation of TSH level above 0.5 mU/L was observed post baseline in 57% of lenvatinib-treated patients as compared with 14% of placebo-treated patients.
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), hypothyroidism occurred in 24% of patients in the lenvatinib plus everolimus-treated group and 2% of patients in the everolimus-treated group. All events of hypothyroidism in the lenvatinib plus everolimus-treated group were of Grade 1 or 2. In patients with a normal TSH at baseline, an elevation of TSH level was observed post baseline in 60.5% of lenvatinib plus everolimus-treated patients as compared with none in patients receiving everolimus alone.
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), 89.6% of patients had a baseline TSH level of less than the upper limit of normal. Elevation of TSH above the upper limit of normal was observed post baseline in 69.6% of lenvatinib-treated patients.
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), hypothyroidism was reported in 68.2% of lenvatinib plus pembrolizumab-treated patients and Grade ≥3 reactions occurred in 1.2% of patients. The median time to onset was 62.0 days. Dose interruption and reduction of lenvatinib occurred in 2.2% and 0.7% of patients, respectively.
Blood TSH increased was reported in 12.8% of lenvatinib plus pembrolizumab-treated patients with no patients reporting Grade ≥3 reactions. Dose interruption occurred in 0.2% of patients.
Diarrhoea (see Precautions): In the DTC study (see Pharmacology: Pharmacodynamics under Actions), diarrhoea was reported in 67.4% of patients in the lenvatinib-treated group (9.2% were Grade ≥3) and in 16.8% of patients in the placebo group (none were Grade ≥3).
In RCC Study 205 (see Pharmacology: Pharmacodynamics under Actions), diarrhoea was reported in 80.6% of patients in the lenvatinib plus everolimus-treated group (21.0% were Grade ≥3) and in 34.0% of patients in the everolimus-treated group (2.0% were Grade ≥3). The median time to onset was 4.1 weeks (any grade) and 8.1 weeks (Grade ≥3) in the lenvatinib plus everolimus-treated group. Diarrhoea was the most frequent cause of dose interruption/reduction and recurred despite dose reduction. Diarrhoea resulted in discontinuation in one patient.
In the HCC study (see Pharmacology: Pharmacodynamics under Actions), diarrhoea was reported in 38.7% of patients treated with lenvatinib (4.2% were Grade ≥3).
In the EC Study 309 (see Pharmacology: Pharmacodynamics under Actions), diarrhoea was reported in 54.2% of lenvatinib plus pembrolizumabtreated patients (7.6% were Grade ≥3). Dose interruption, reduction and discontinuation of lenvatinib occurred in 10.6%, 11.1% and 1.2% of patients, respectively.
Paediatric population: Clinical data are not yet available in this population (see Dosage & Administration).
Other special populations: Elderly: In CLEAR, patients of age ≥75 years had a higher (≥10% difference) incidence of proteinuria than patients of age <65 years.
In DTC, patients of age ≥75 years were more likely to experience Grade 3 or 4 hypertension, proteinuria, decreased appetite, and dehydration.
In RCC study, there are limited data on patients of age ≥75 years with RCC.
In HCC, patients of age ≥75 years were more likely to experience hypertension, proteinuria, decreased appetite, asthenia, dehydration, dizziness, malaise, peripheral oedema, pruritus and hepatic encephalopathy. Hepatic encephalopathy occurred at more than twice the incidence in patients aged ≥75 years (17.2%) than in those <75 years (7.1%). Hepatic encephalopathy tended to be associated with adverse disease characteristics at baseline or with the use of concomitant medications. Arterial thromboembolic events also occurred at an increased incidence in this age group.
In EC, patients of age ≥75 years were more likely to experience urinary tract infections and Grade ≥3 hypertension (≥10% increase compared to patients of age <65 years).
Gender: In CLEAR, males had a higher (≥10% difference) incidence than females of diarrhoea.
In patients with DTC, females had a higher incidence of hypertension (including Grade 3 or 4 hypertension), proteinuria, and PPE, while males had a higher incidence of decreased ejection fraction and gastrointestinal perforation and fistula formation.
In HCC, females had a higher incidence of hypertension, fatigue, ECG QT prolongation and alopecia. Men had a higher incidence (26.5%) of dysphonia than women (12.3%), decreased weight and decreased platelet count. Hepatic failure events were observed in male patients only.
Ethnic origin: In CLEAR, Asian patients had a higher (≥10% difference) incidence than Caucasian patients of palmar-plantar erythrodysaesthesia syndrome, proteinuria and hypothyroidism (including blood thyroid hormone increased) while Caucasian patients had a higher incidence of fatigue, nausea, arthralgia, vomiting, and asthenia.
In patients with DTC, Asian patients had a higher (≥10% difference) incidence than Caucasian patients of peripheral oedema, hypertension, fatigue, PPE, proteinuria, stomatitis, thrombocytopenia, and myalgia; while Caucasian patients had a higher incidence of diarrhoea, weight decreased, nausea, vomiting, constipation, asthenia, abdominal pain, pain in extremity, and dry mouth. A larger proportion of Asian patients had a lenvatinib dose reduction compared to Caucasian patients. The median time to first dose reduction and the average daily dose taken were lower in Asian than in Caucasian patients.
In RCC Study 205, there are limited data on Asian patients.
In HCC, Asian patients had a higher incidence than Caucasian patients of proteinuria, decreased neutrophil count, decreased platelet count, decreased white blood count and PPE syndrome, while Caucasian patients had a higher incidence of fatigue, hepatic encephalopathy, acute kidney injury, anxiety, asthenia, nausea, thrombocytopenia and vomiting.
In EC, Asian patients had a higher (≥10% difference) incidence than Caucasian patients of anaemia, malaise, neutrophil count decrease, stomatitis, platelet count decreased, proteinuria and PPE while Caucasian patients had a higher incidence of mucosal inflammation, abdominal pain, diarrhoea, urinary tract infection, weight
decreased, hypomagnesaemia, dizziness, asthenia and fatigue.
Baseline hypertension: In CLEAR, patients with baseline hypertension had a higher incidence of proteinuria than patients without baseline hypertension.
In DTC, patients with baseline hypertension had a higher incidence of Grade 3 or 4 hypertension, proteinuria, diarrhoea, and dehydration, and experienced more serious cases of dehydration, hypotension, pulmonary embolism, malignant pleural effusion, atrial fibrillation, and GI symptoms (abdominal pain, diarrhoea, vomiting).
In RCC Study 205, patients with baseline hypertension had a higher incidence of Grade 3 or 4 dehydration, fatigue, and hypertension.
Baseline diabetes: In RCC Study 205, patients with baseline diabetes had a higher incidence of Grade 3 or 4 hypertension, hypertriglyceridemia and acute renal failure.
Hepatic impairment: In DTC, patients with baseline hepatic impairment had a higher incidence of hypertension and PPE, and a higher incidence of Grade 3 or 4 hypertension, asthenia, fatigue, and hypocalcaemia compared with patients with normal hepatic function.
In RCC Study 205, there are limited data on patients with hepatic impairment.
In HCC, patients with a baseline Child Pugh (CP) score of 6 (about 20% patients in the REFLECT study) had a higher incidence of decreased appetite, fatigue, proteinuria, hepatic encephalopathy and hepatic failure compared to patients with a baseline CP score of 5. Hepatotoxicity events and haemorrhage events also occurred at a higher incidence in CP score 6 patients compared to CP score 5 patients.
Renal impairment: In DTC, patients with baseline renal impairment had a higher incidence of Grade 3 or 4 hypertension, proteinuria, fatigue, stomatitis, oedema peripheral, thrombocytopenia, dehydration, prolonged electrocardiogram QT, hypothyroidism, hyponatraemia, blood thyroid stimulating hormone increased, pneumonia compared with subjects with normal renal function. These patients also had a higher incidence of renal reactions and a trend towards a higher incidence of liver reactions.
In RCC Study 205, patients with baseline renal impairment had a higher incidence of Grade 3 fatigue.
In HCC, patients with baseline renal impairment had a higher incidence of fatigue, hypothyroidism, dehydration, diarrhoea, decreased appetite, proteinuria and hepatic encephalopathy. These patients also had a higher incidence of renal reactions and arterial thromboembolic events.
Patients with body weight <60 kg: In DTC, patients with low body weight (<60 kg) had a higher incidence of PPE, proteinuria, of Grade 3 or 4 hypocalcaemia and hyponatraemia, and a trend towards a higher incidence of Grade 3 or 4 decreased appetite. There are limited data on patients with body weight <60 kg in RCC.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image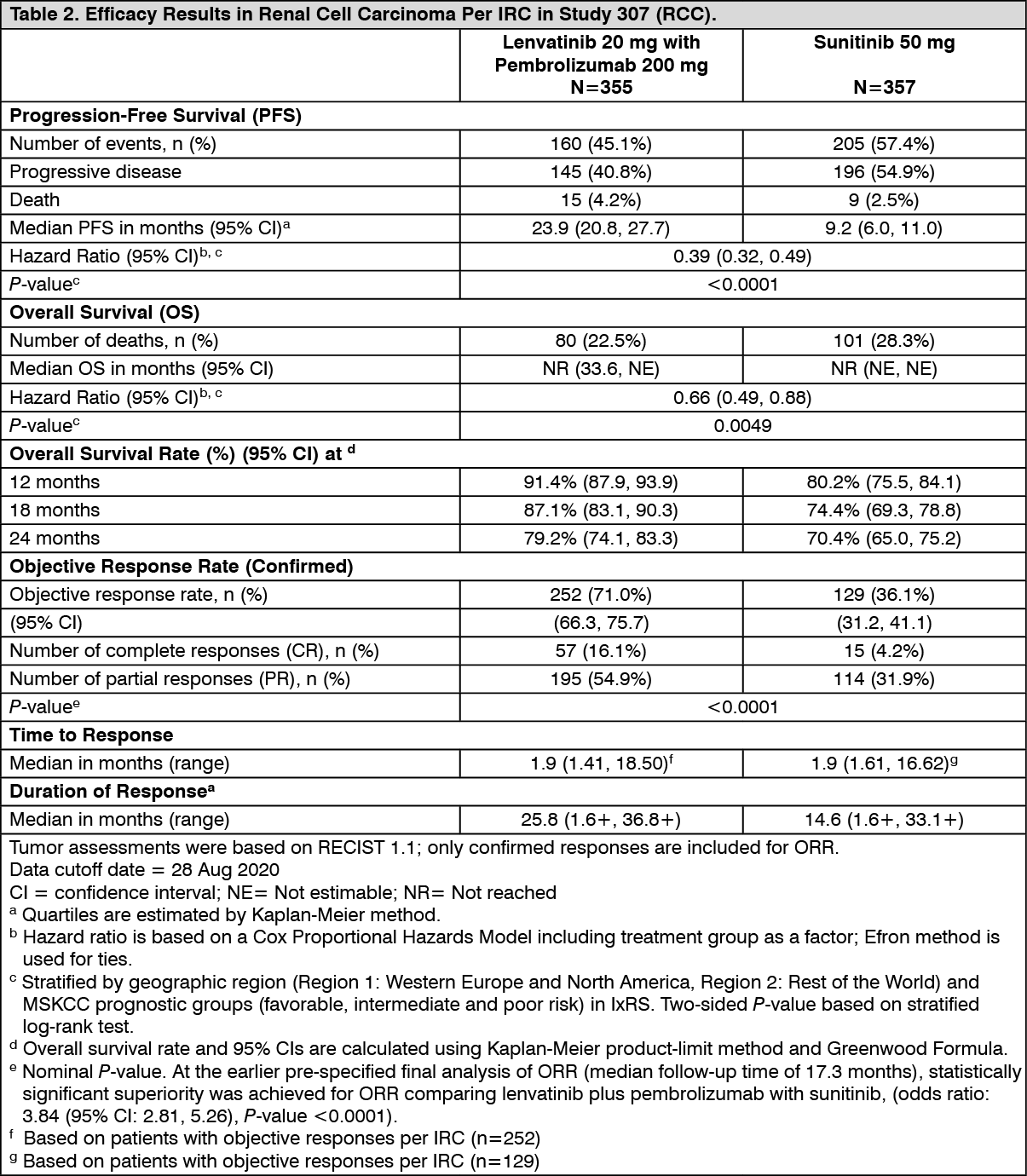 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image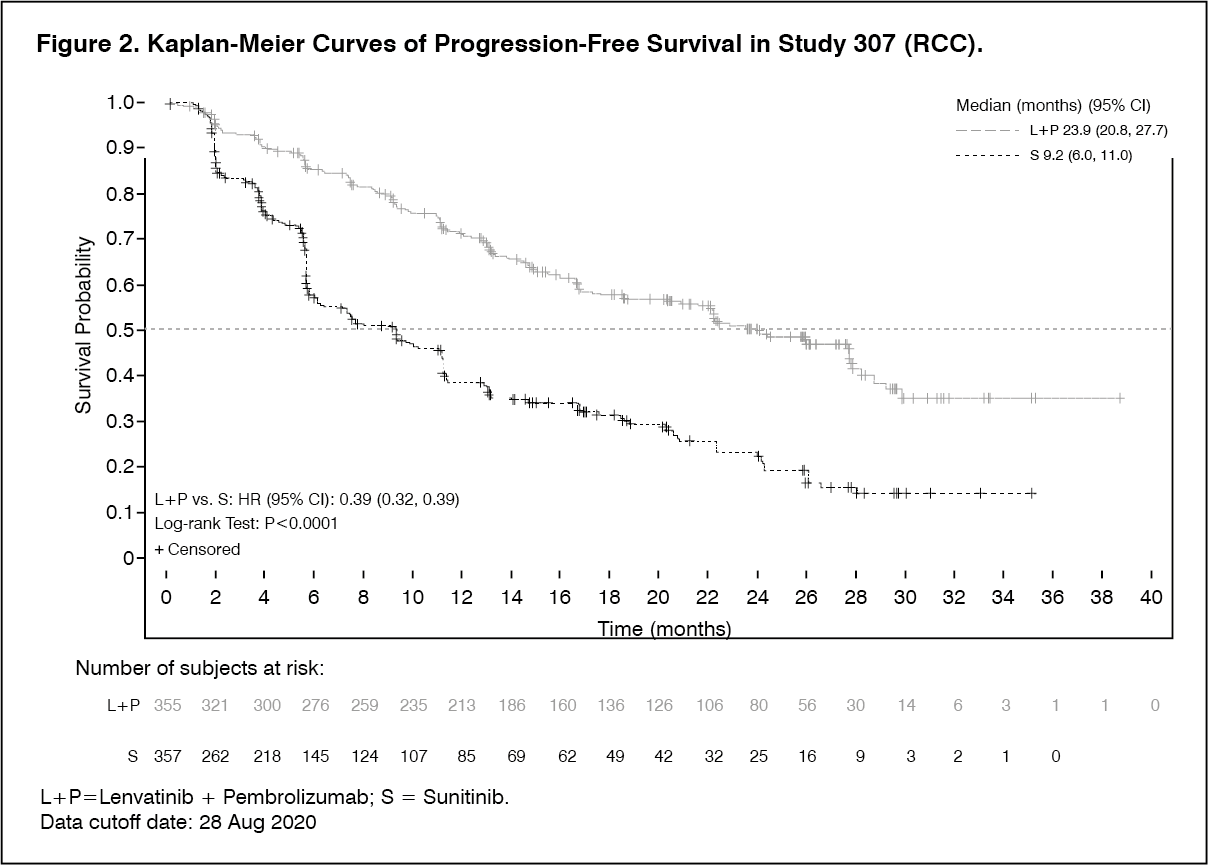 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image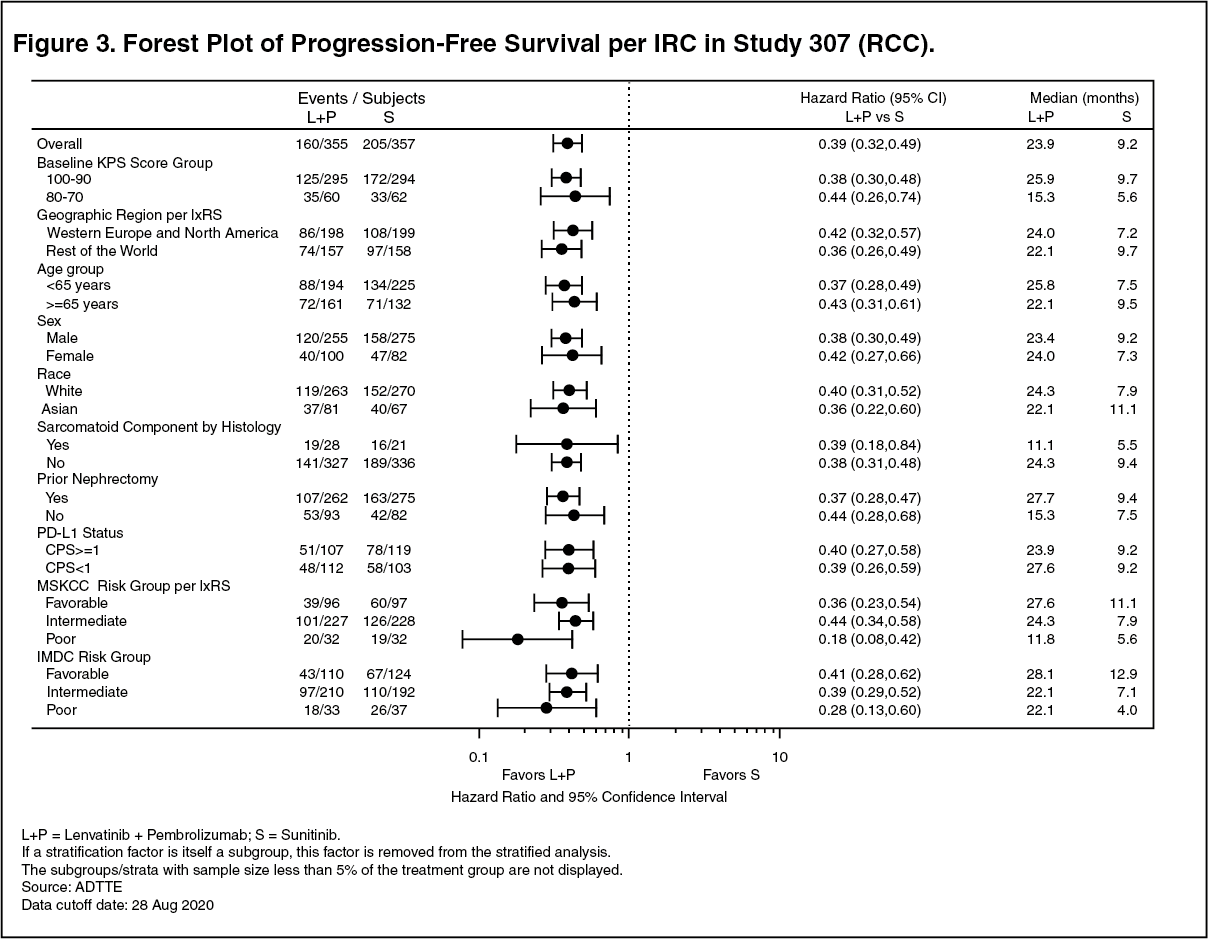 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image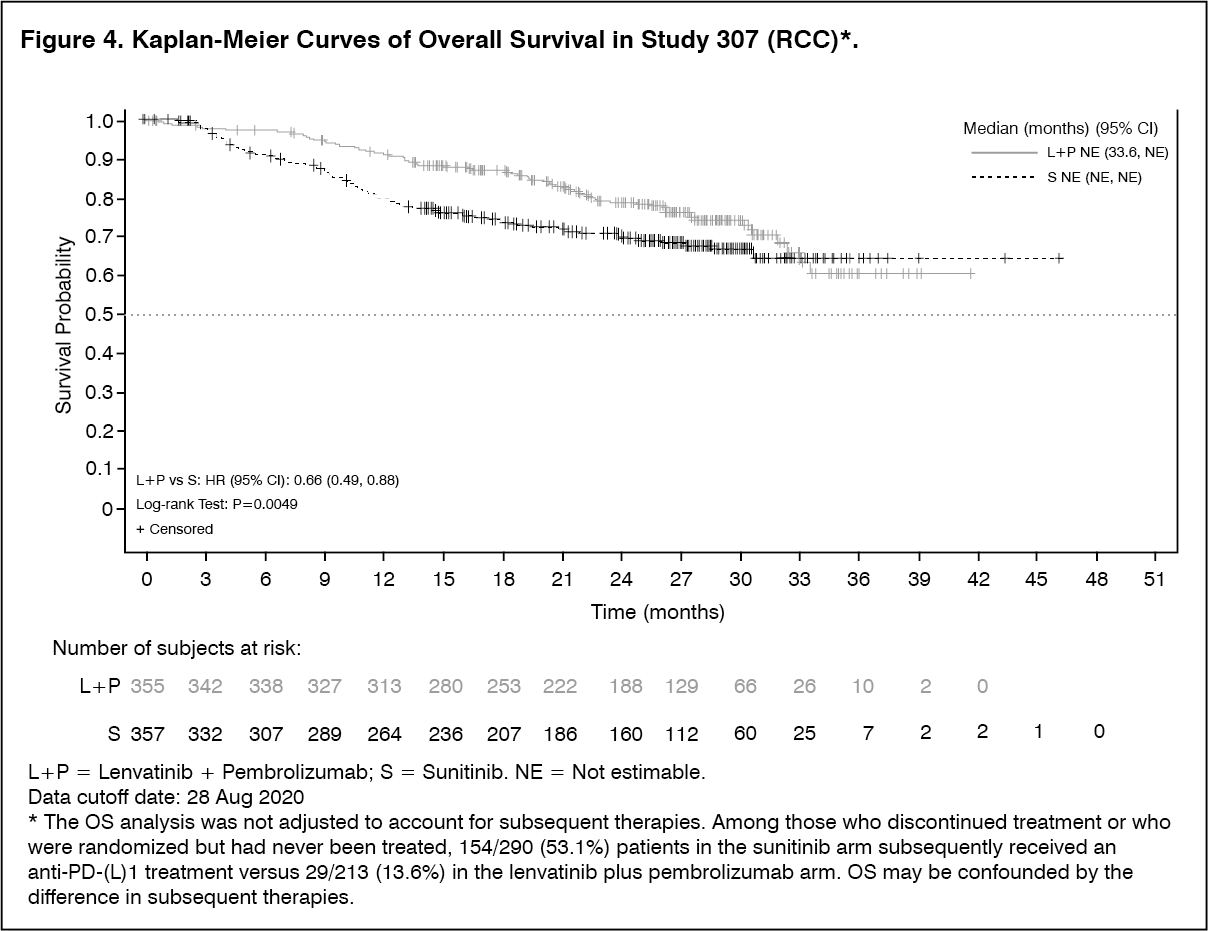 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image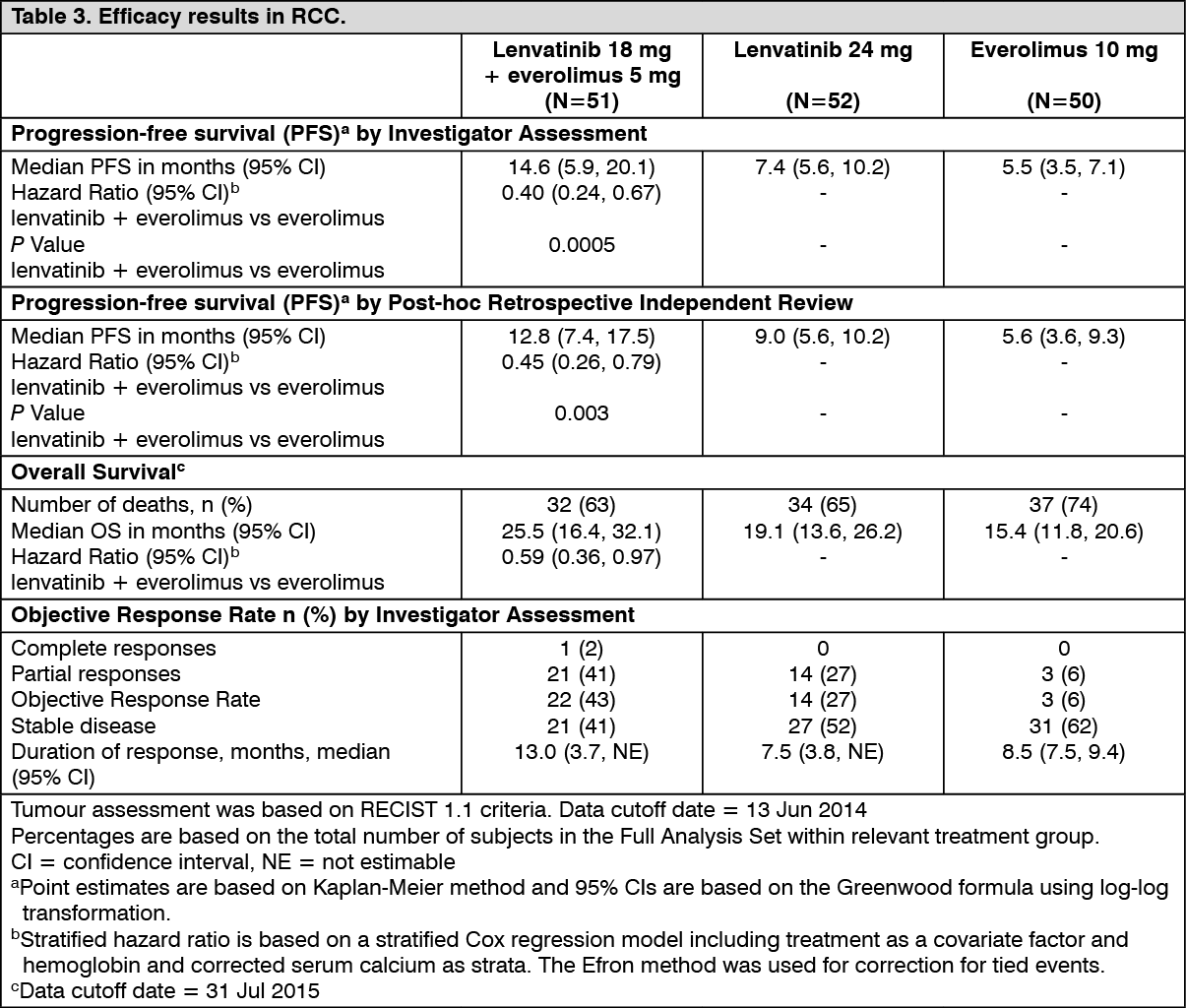 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image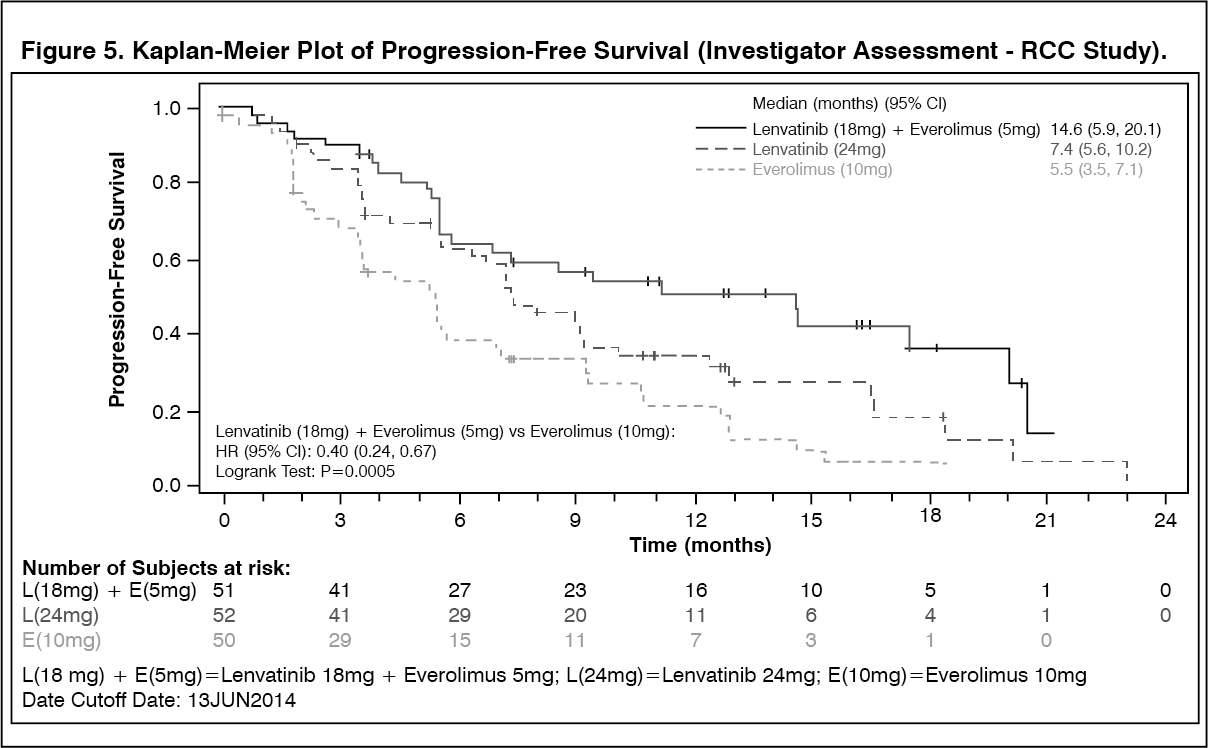 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image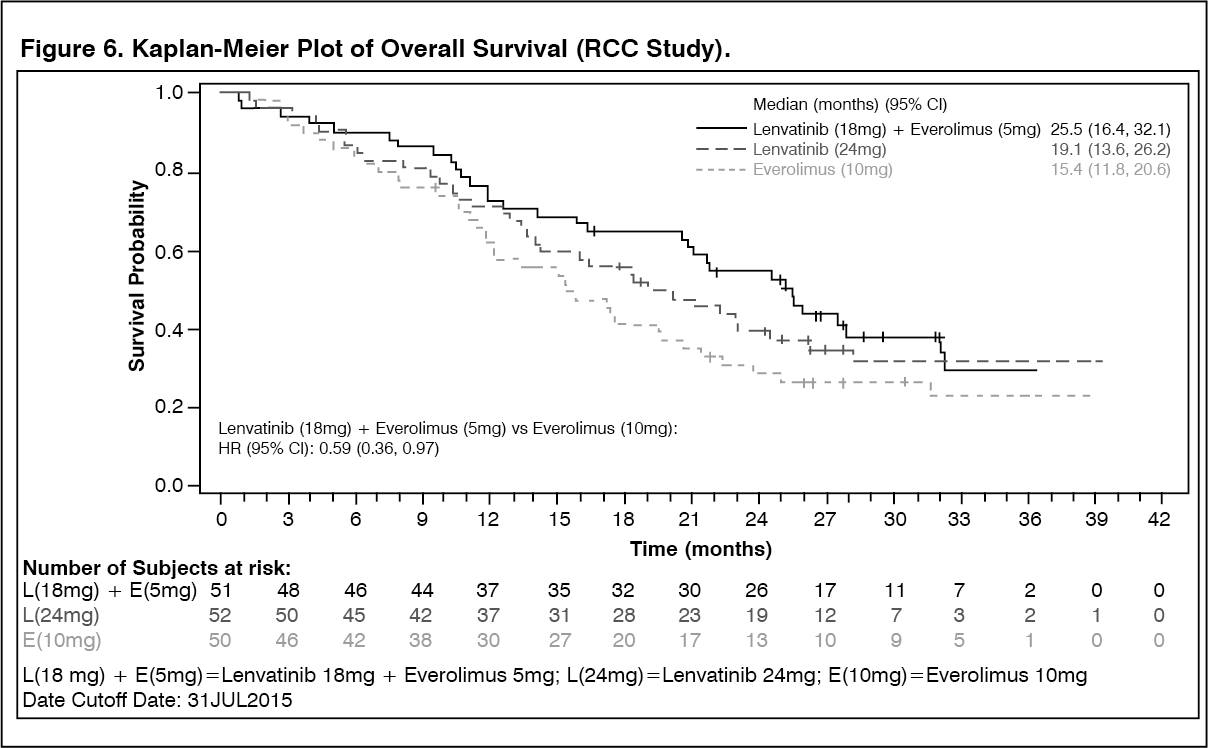 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image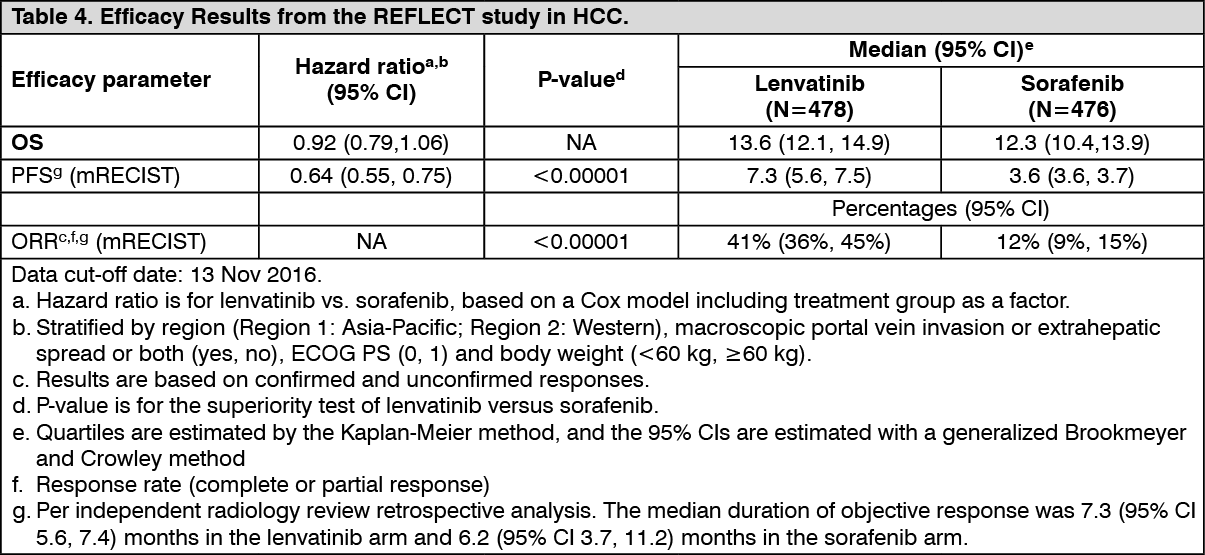 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image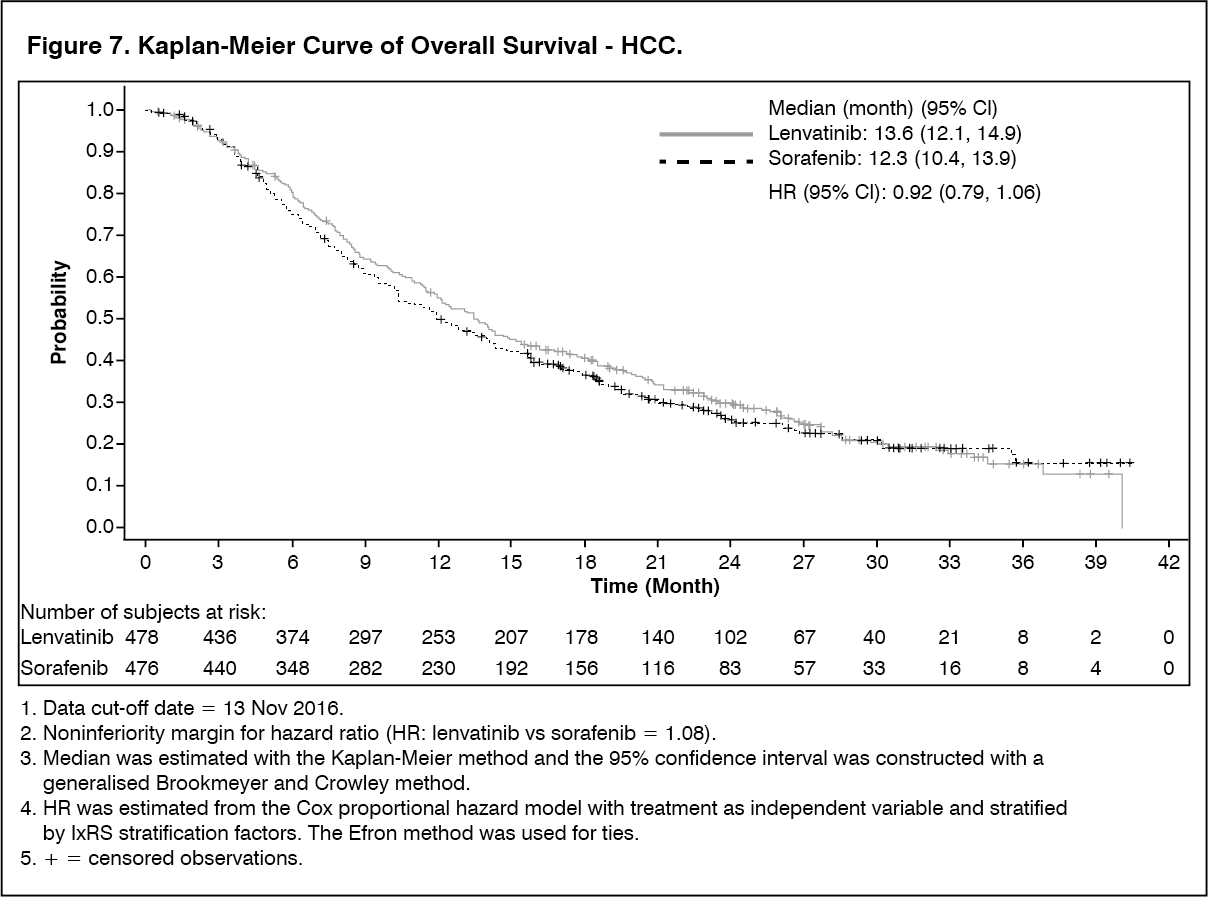 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image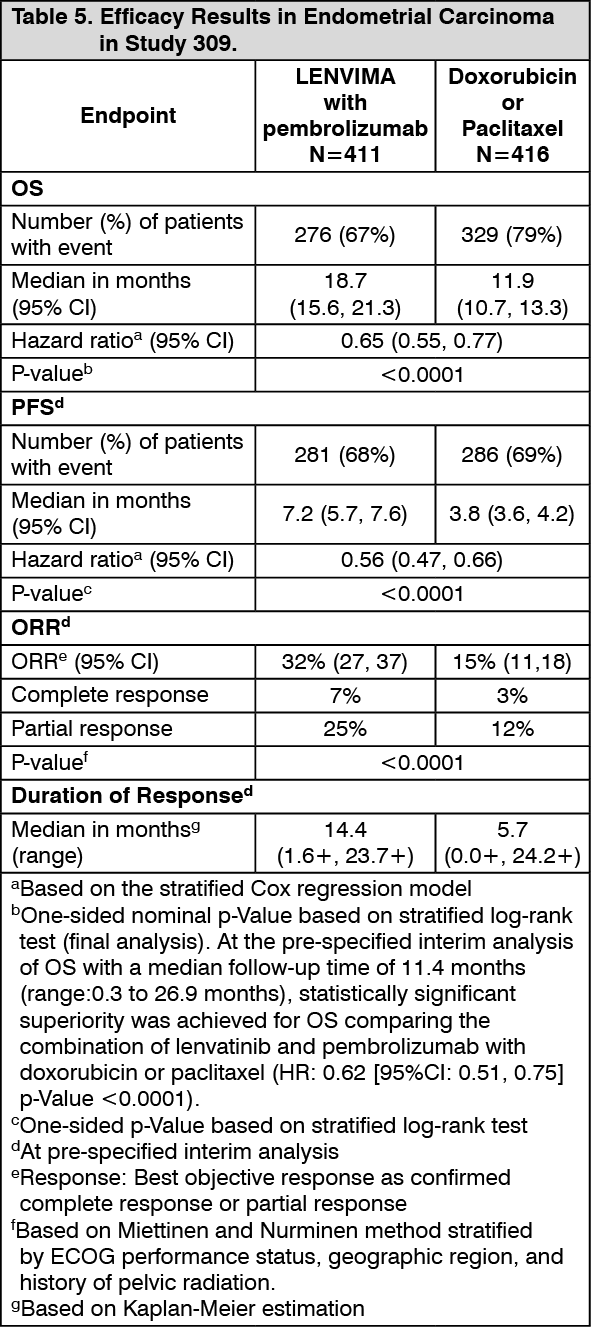 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image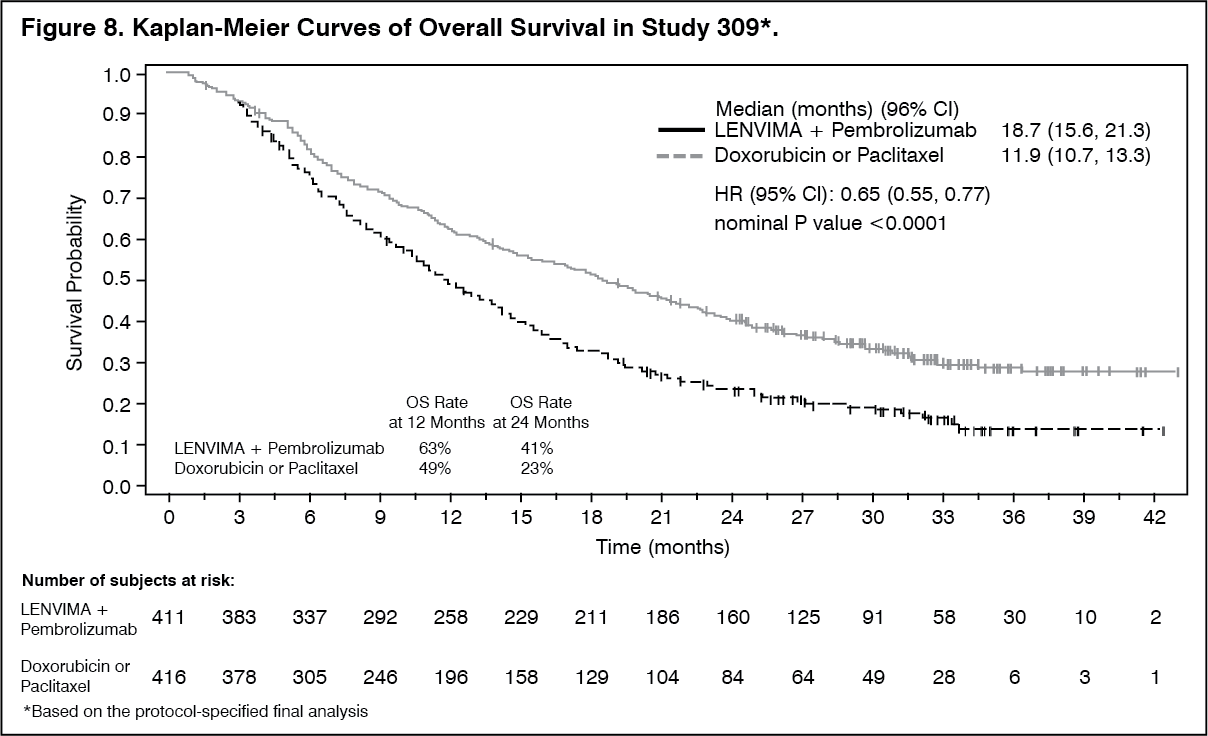 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image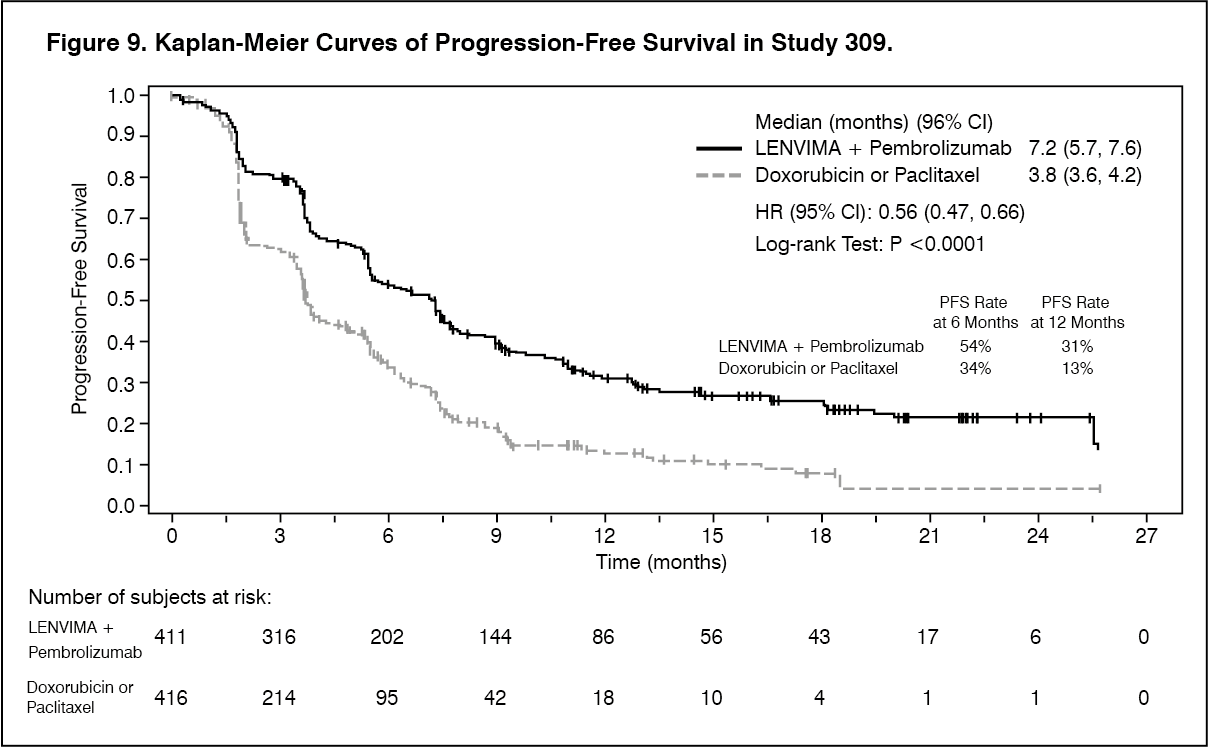 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image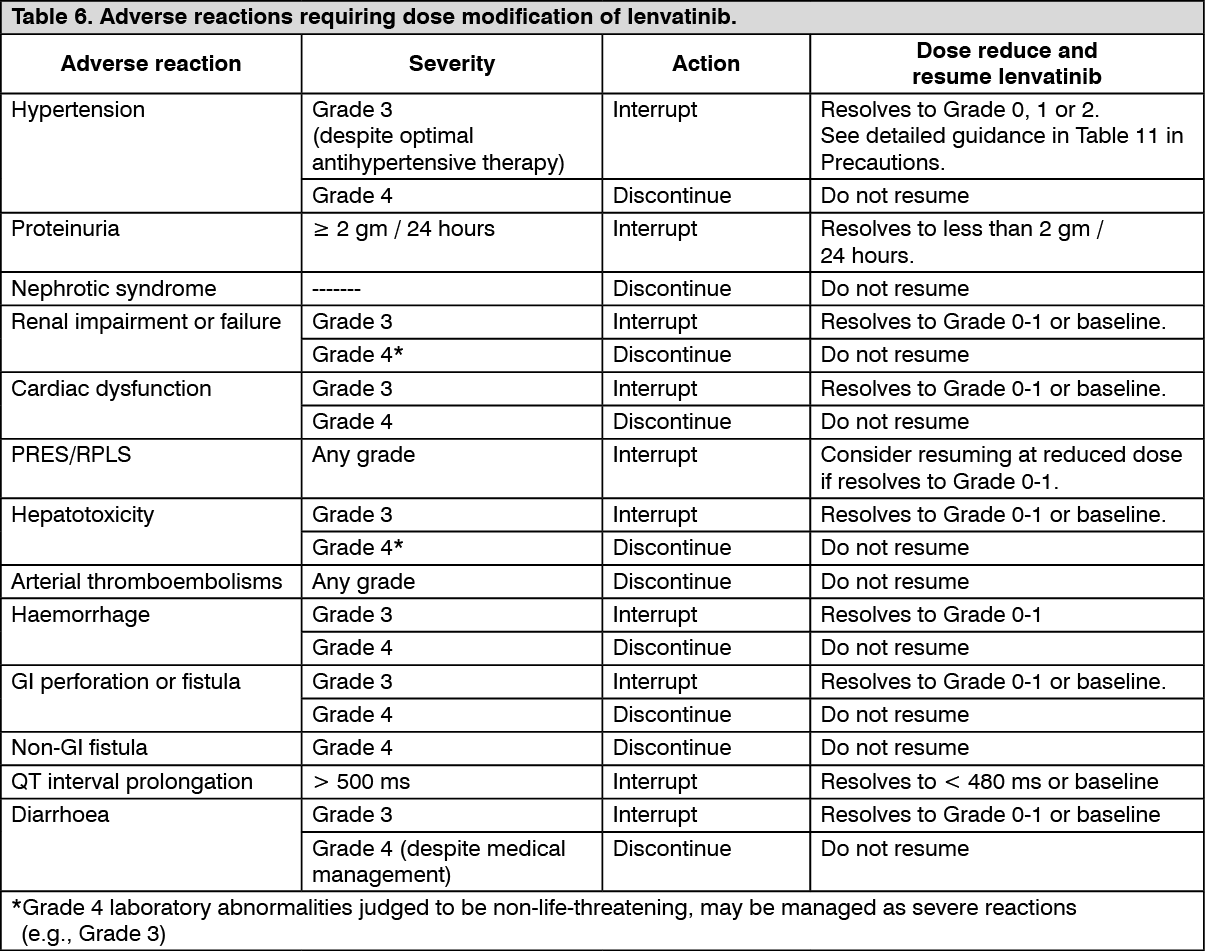 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image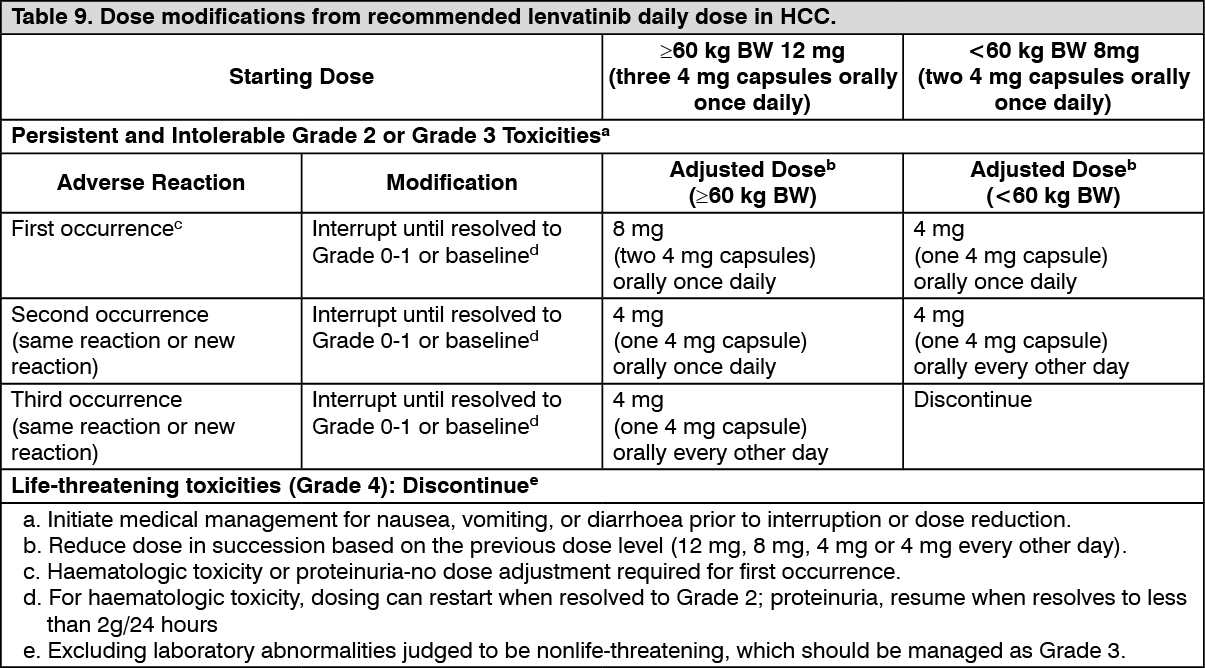 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image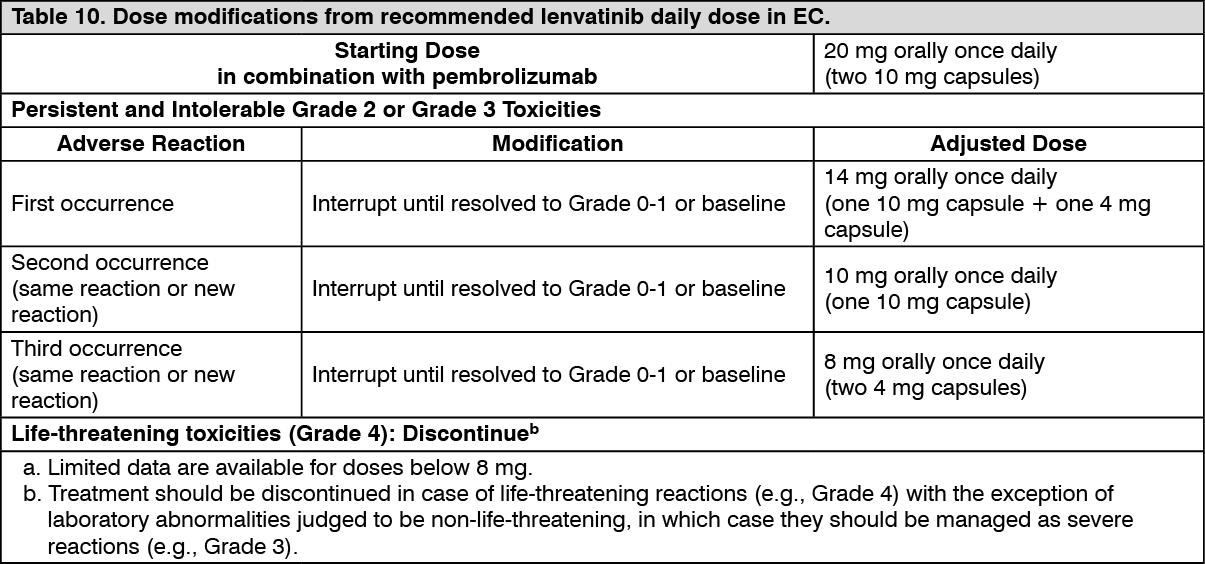 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image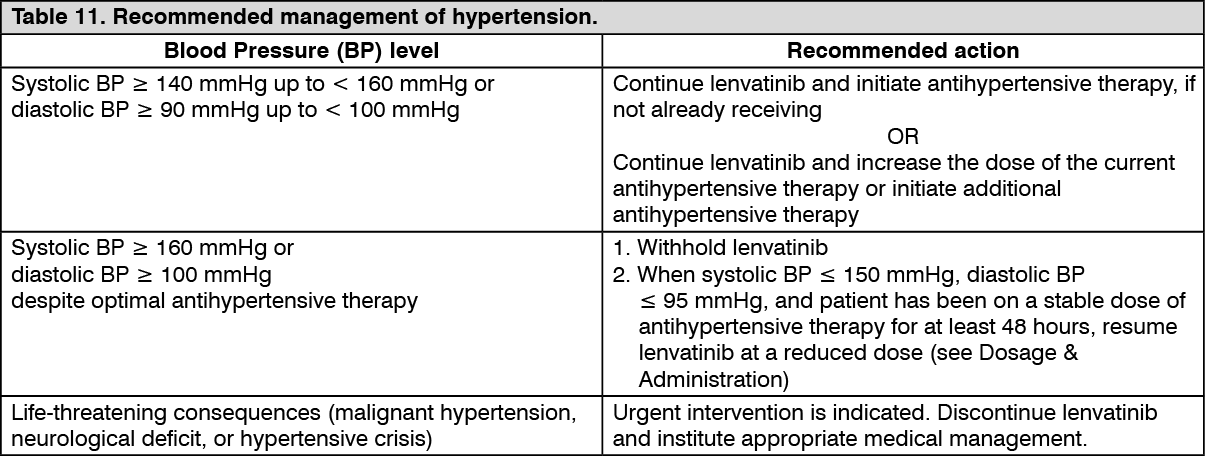 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image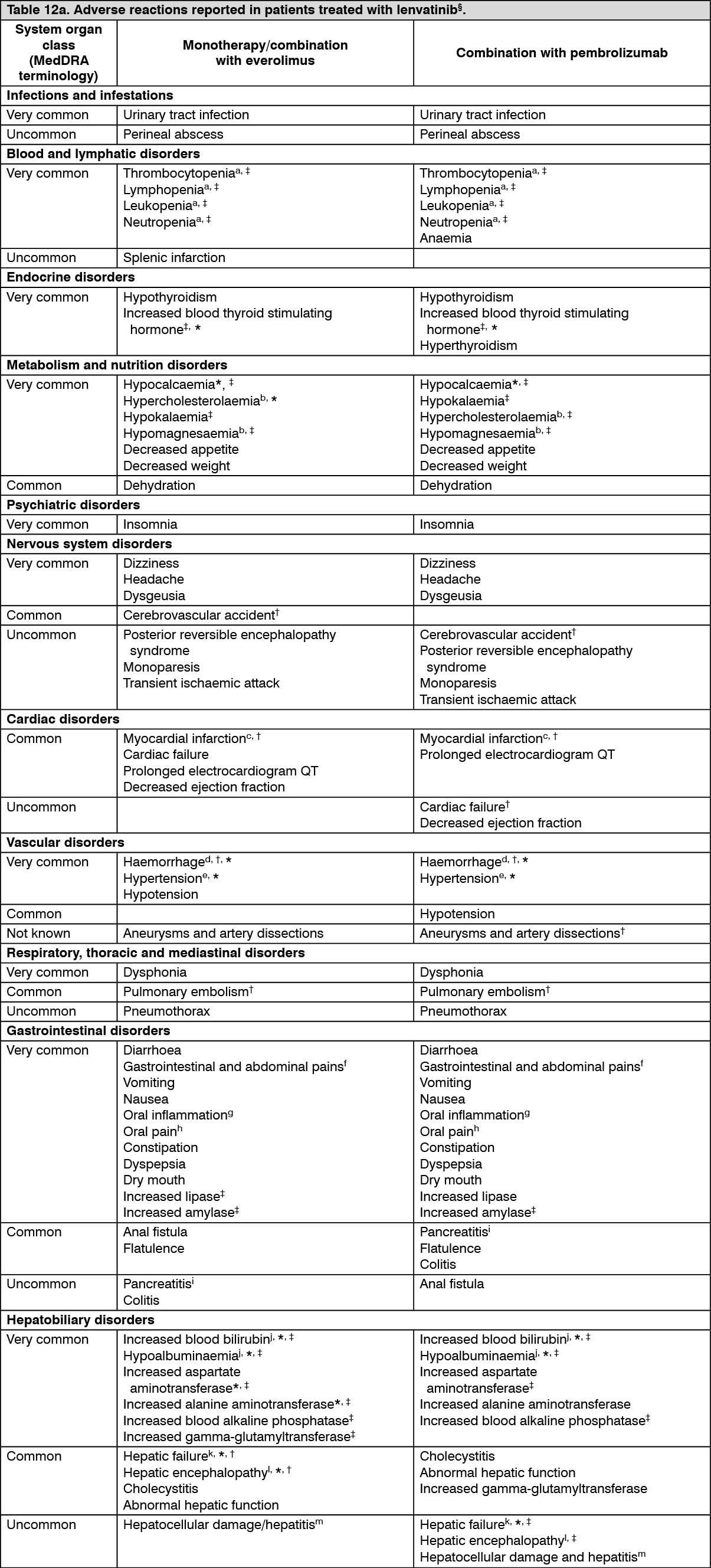 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image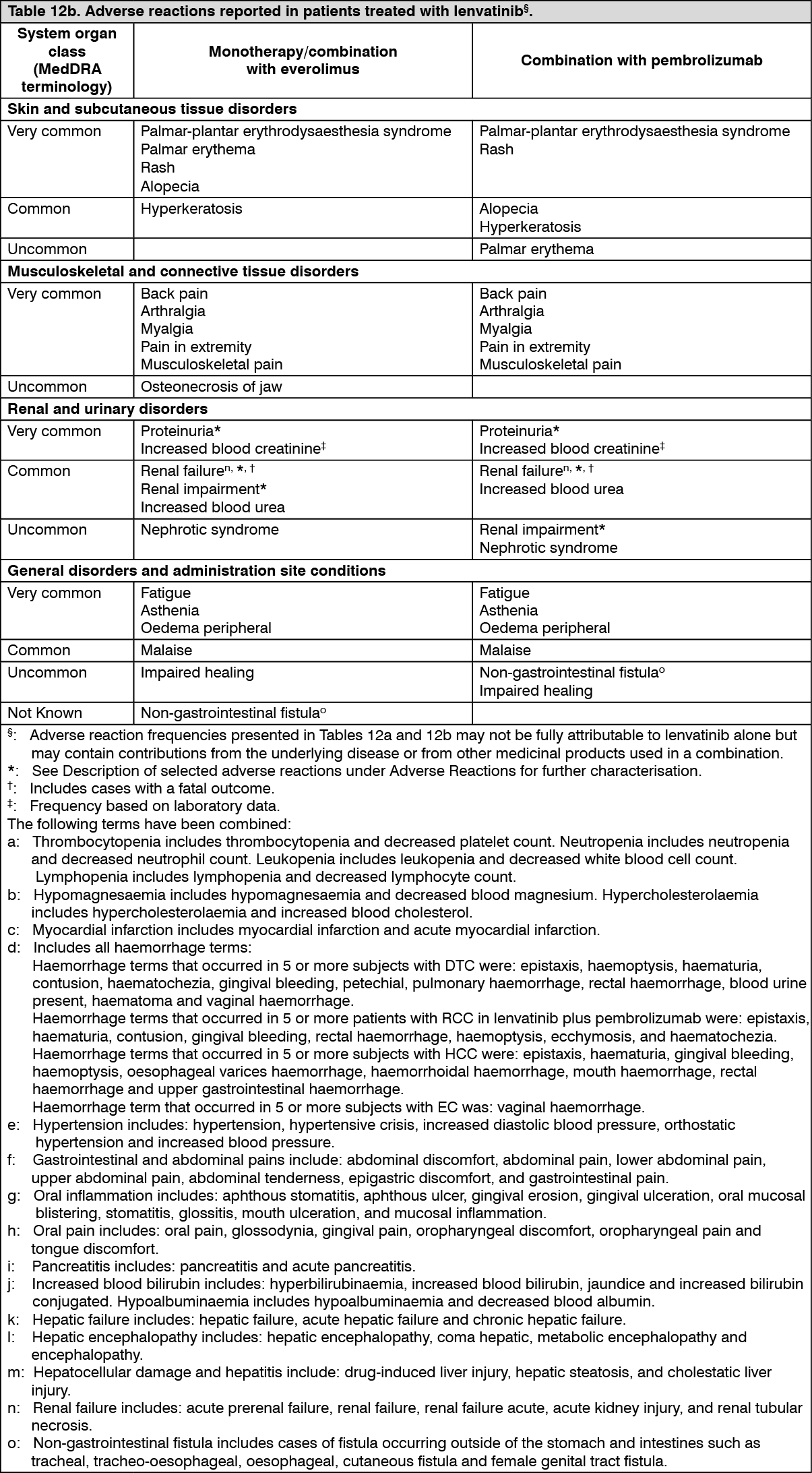 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
