


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Axitinib is a potent and selective tyrosine kinase inhibitor of vascular endothelial growth factor receptors (VEGFR)-1, VEGFR-2 and VEGFR-3. These receptors are implicated in pathologic angiogenesis, tumour growth, and metastatic progression of cancer. Axitinib has been shown to potently inhibit VEGF-mediated endothelial cell proliferation and survival. Axitinib inhibited the phosphorylation of VEGFR-2 in xenograft tumour vasculature that expressed the target in vivo and produced tumour growth delay, regression and inhibition of metastases in many experimental models of cancer.
Effect on QTc interval: In a randomised, 2-way crossover study, 35 healthy subjects were administered a single oral dose of axitinib (5 mg) in the absence and presence of 400 mg ketoconazole for 7 days. Results of this study indicated that axitinib plasma exposures up to two-fold greater than therapeutic levels expected following a 5 mg dose, did not produce clinically-significant QT interval prolongation.
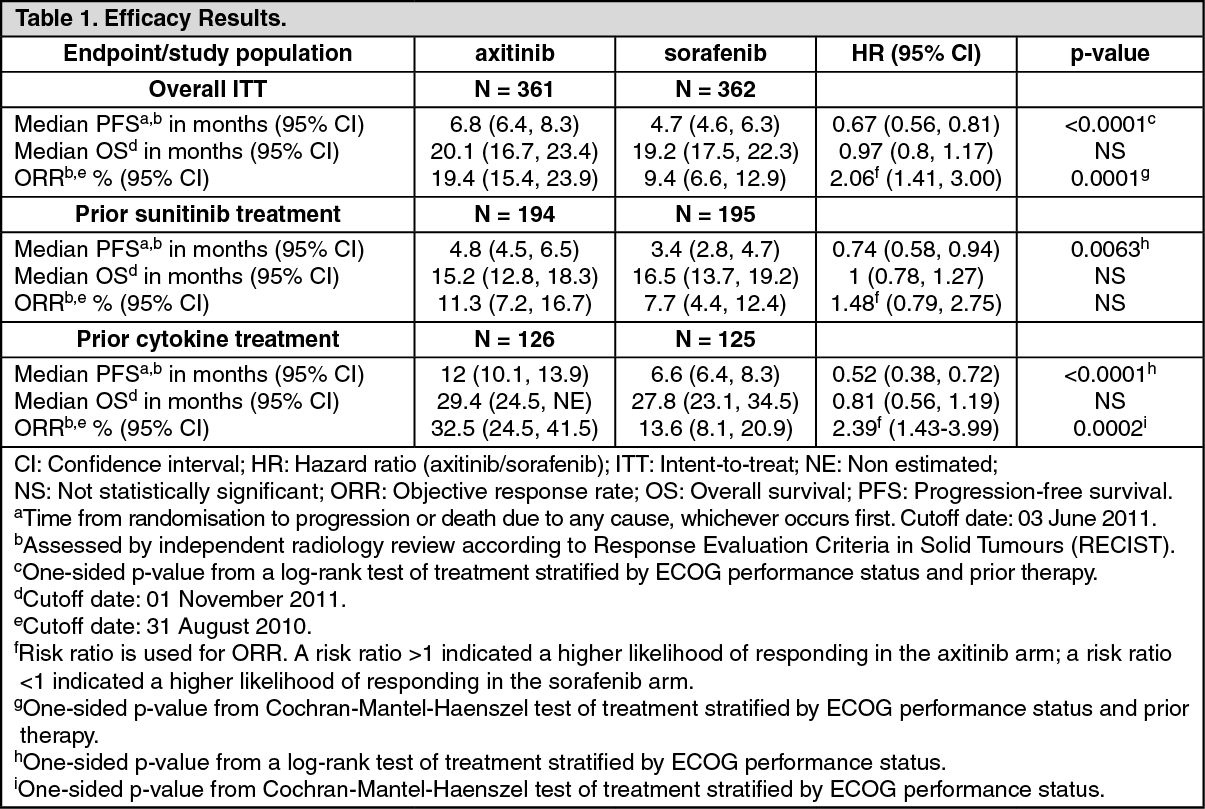
Clinical efficacy and safety: The safety and efficacy of axitinib were evaluated in a randomised, open-label, multicenter Phase 3 study. Patients (N = 723) with advanced RCC whose disease had progressed on or after treatment with one prior systemic therapy, including sunitinib-, bevacizumab-, temsirolimus-, or cytokine-containing regimens were randomised (1:1) to receive axitinib (N = 361) or sorafenib (N = 362). The primary endpoint, progression-free survival (PFS), was assessed using a blinded independent central review. Secondary endpoints included objective response rate (ORR) and overall survival (OS).
Of the patients enrolled in this study, 389 patients (53.8%) had received one prior sunitinib-based therapy, 251 patients (34.7%) had received one prior cytokine-based therapy (interleukin-2 or interferon-alpha), 59 patients (8.2%) had received one prior bevacizumab-based therapy, and 24 patients (3.3%) had received one prior temsirolimus-based therapy. The baseline demographic and disease characteristics were similar between the axitinib and sorafenib groups with regard to age, gender, race, Eastern Cooperative Oncology Group (ECOG) performance status, geographic region, and prior treatment.
In the overall patient population and the two main subgroups (prior sunitinib treatment and prior cytokine treatment), there was a statistically significant advantage for axitinib over sorafenib for the primary endpoint of PFS (see Table 1 and Figures 1, 2 and 3). The magnitude of median PFS effect was different in the subgroups by prior therapy. Two of the subgroups were too small to give reliable results (prior temsirolimus treatment or prior bevacizumab treatment). There were no statistically significant differences between the arms in OS in the overall population or in the subgroups by prior therapy. (See Table 1 and Figures 1, 2 and 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image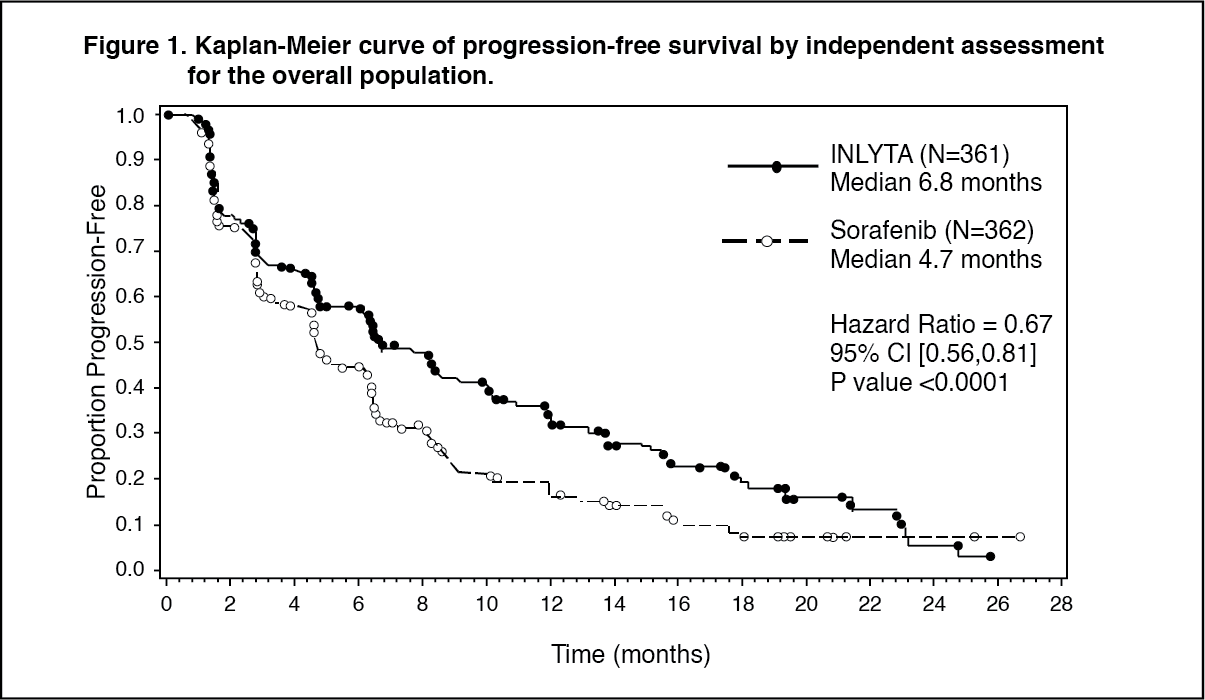 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image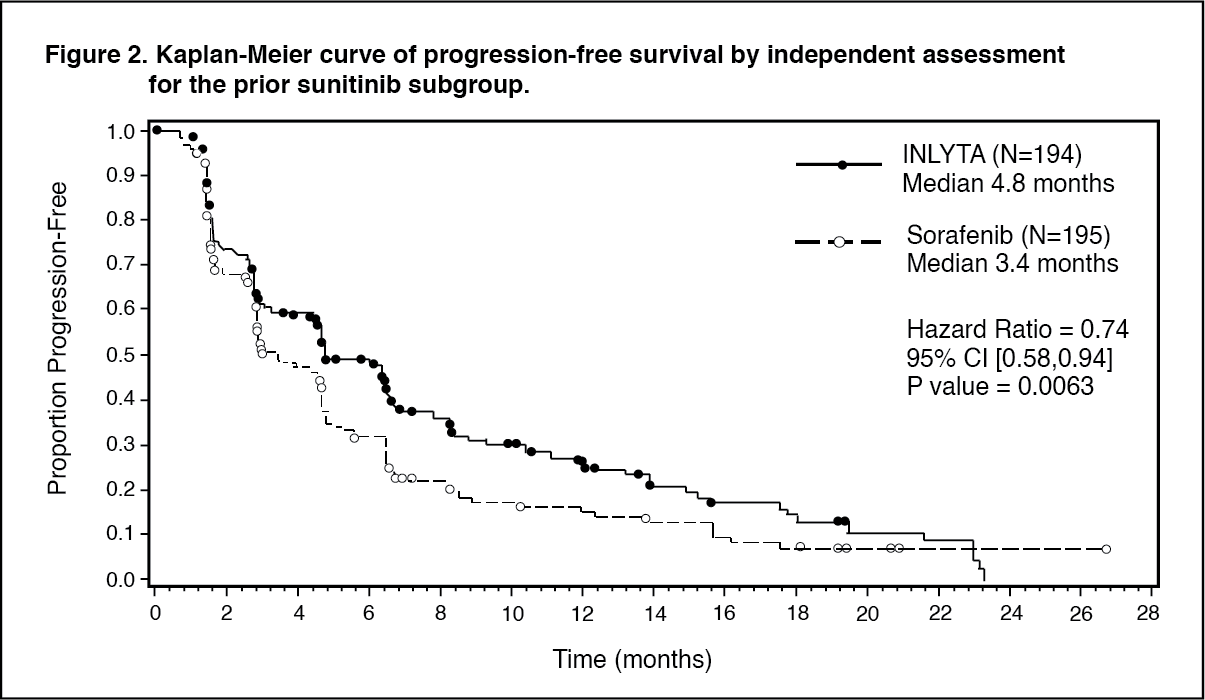 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePaediatric population: The European Medicines Agency has waived the obligation to submit the results of studies with axitinib in all subsets of the paediatric population for treatment of kidney and renal pelvis carcinoma (excluding nephroblastoma, nephroblastomatosis, clear cell sarcoma, mesoblastic nephroma, renal medullary carcinoma and rhabdoid tumour of the kidney) (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: After oral administration of axitinib tablets, the mean absolute bioavailability is 58% compared to intravenous administration. The plasma half-life of axitinib ranges from 2.5 to 6.1 hours. Dosing of axitinib at 5 mg twice daily resulted in less than two-fold accumulation compared to administration of a single dose. Based on the short half-life of axitinib, steady-state is expected within 2 to 3 days of the initial dose.
Absorption and distribution: Peak axitinib concentrations in plasma are generally reached within 4 hours following oral administration of axitinib with median Tmax ranging from 2.5 to 4.1 hours. Administration of axitinib with a moderate fat meal resulted in 10% lower exposure compared to overnight fasting. A high-fat, high-calorie meal resulted in 19% higher exposure compared to overnight fasting. Axitinib may be administered with or without food (see Dosage & Administration).
The average Cmax and AUC increased proportionally over an axitinib dosing range of 5 to 10 mg. In vitro binding of axitinib to human plasma proteins is >99% with preferential binding to albumin and moderate binding to α1-acid glycoprotein. At the 5 mg twice daily dose in the fed state, the geometric mean peak plasma concentration and 24-hour AUC were 27.8 ng/mL and 265 ng·h/mL, respectively, in patients with advanced RCC. The geometric mean oral clearance and apparent volume of distribution were 38 L/h and 160 L, respectively.
Biotransformation and elimination: Axitinib is metabolised primarily in the liver by CYP3A4/5 and to a lesser extent by CYP1A2, CYP2C19, and UGT1A1.
Following oral administration of a 5 mg radioactive dose of axitinib, 30-60% of the radioactivity was recovered in faeces and 23% of the radioactivity was recovered in urine. Unchanged axitinib, accounting for 12% of the dose, was the major component identified in faeces. Unchanged axitinib was not detected in urine; the carboxylic acid and sulfoxide metabolites accounted for the majority of radioactivity in urine. In plasma, the N-glucuronide metabolite represented the predominant radioactive component (50% of circulating radioactivity) and unchanged axitinib and the sulfoxide metabolite each accounted for approximately 20% of the circulating radioactivity.
The sulfoxide and N-glucuronide metabolites show approximately 400-fold and 8000-fold less in vitro potency, respectively, against VEGFR-2 compared to axitinib.
Special populations: Elderly, gender, and race: Population pharmacokinetic analyses in patients with advanced cancer (including advanced RCC) and healthy volunteers indicate that there are no clinically relevant effects of age, gender, body weight, race, renal function, UGT1A1 genotype, or CYP2C19 genotype.
Paediatric population: Axitinib has not been studied in patients <18 years of age.
Hepatic impairment: In vitro and in vivo data indicate that axitinib is primarily metabolised by the liver.
Compared to subjects with normal hepatic function, systemic exposure following a single dose of axitinib was similar in subjects with mild hepatic impairment (Child-Pugh class A) and higher (approximately two-fold) in subjects with moderate hepatic impairment (Child-Pugh class B). Axitinib has not been studied in subjects with severe hepatic impairment (Child-Pugh class C) and should not be used in this population (see Dosage & Administration for dose adjustment recommendations).
Renal impairment: Unchanged axitinib is not detected in the urine.
Axitinib has not been studied in subjects with renal impairment. In clinical studies with axitinib for the treatment of patients with RCC, patients with serum creatinine >1.5 times the ULN or calculated creatinine clearance <60 mL/min were excluded. Population pharmacokinetic analyses have shown that axitinib clearance was not altered in subjects with renal impairment and no dose adjustment of axitinib is required.
Toxicology: Preclinical safety data: Repeat dose toxicity: Major toxicity findings in mice and dogs following repeated dosing for up to 9 months were the gastrointestinal, haematopoietic, reproductive, skeletal and dental systems, with No Observed Adverse Effect Levels (NOAEL) approximately equivalent to or below expected human exposure at the recommended clinical starting dose (based on AUC levels).
Carcinogenicity: Carcinogenicity studies have not been performed with axitinib.
Genotoxicity: Axitinib was not mutagenic or clastogenic in conventional genotoxicity assays in vitro. A significant increase in polyploidy was observed in vitro at concentrations >0.22 μg/mL, and an elevation in micronucleated polychromatic erythrocytes was observed in vivo with No Observed Effect Level (NOEL) 69-fold the expected human exposure. Genotoxicity findings are not considered clinically relevant at exposure levels observed in humans.
Reproduction toxicity: Axitinib-related findings in the testes and epididymis included decreased organ weight, atrophy or degeneration, decreased numbers of germinal cells, hypospermia or abnormal sperm forms, and reduced sperm density and count. These findings were observed in mice at exposure levels approximately 12-fold the expected human exposure, and in dogs at exposure levels below the expected human exposure. There was no effect on mating or fertility in male mice at exposure levels approximately 57-fold the expected human exposure. Findings in females included signs of delayed sexual maturity, reduced or absent corpora lutea, decreased uterine weights and uterine atrophy at exposures approximately equivalent to the expected human exposure. Reduced fertility and embryonic viability were observed in female mice at all doses tested, with exposure levels at the lowest dose approximately 10-fold the expected human exposure.
Pregnant mice exposed to axitinib showed an increased occurrence of cleft palate malformations and skeletal variations, including delayed ossification, at exposure levels below the expected human exposure. Perinatal and postnatal developmental toxicity studies have not been conducted.
Toxicity findings in immature animals: Reversible physeal dysplasia was observed in mice and dogs given axitinib for at least 1 month at exposure levels approximately six-fold higher than the expected human exposure. Partially reversible dental caries were observed in mice treated for more than 1 month at exposure levels similar to the expected human exposure. Other toxicities of potential concern to paediatric patients have not been evaluated in juvenile animals.