Pharmacotherapeutic group: Drugs used in diabetes, dipeptidyl peptidase 4 (DPP-4) inhibitors.
ATC code: A10BH02.
Pharmacology: Pharmacodynamics: Vildagliptin, a member of the islet enhancer class, is a potent and selective DPP-4 inhibitor.
Mechanism of action: The administration of vildagliptin results in a rapid and complete inhibition of DPP-4 activity, resulting in increased fasting and postprandial endogenous levels of the incretin hormones GLP-1 (glucagon-like peptide 1) and GIP (glucose-dependent insulinotropic polypeptide).
Pharmacodynamic effects: By increasing the endogenous levels of these incretin hormones, vildagliptin enhances the sensitivity of beta cells to glucose, resulting in improved glucose-dependent insulin secretion. Treatment with vildagliptin 50-100 mg daily in patients with type 2 diabetes significantly improved markers of beta cell function including HOMA-β (Homeostasis Model Assessment-β), proinsulin to insulin ratio and measures of beta cell responsiveness from the frequently-sampled meal tolerance test. In non-diabetic (normal glycaemic) individuals, vildagliptin does not stimulate insulin secretion or reduce glucose levels.
By increasing endogenous GLP-1 levels, vildagliptin also enhances the sensitivity of alpha cells to glucose, resulting in more glucose-appropriate glucagon secretion.
The enhanced increase in the insulin/glucagon ratio during hyperglycaemia due to increased incretin hormone levels results in a decrease in fasting and postprandial hepatic glucose production, leading to reduced glycaemia.
The known effect of increased GLP-1 levels delaying gastric emptying is not observed with vildagliptin treatment.
Clinical efficacy and safety: More than 15000 patients with type 2 diabetes participated in double-blind placebo- or active-controlled clinical trials of up to more than 2 years' treatment duration. In these studies, vildagliptin was administered to more than 9000 patients at daily doses of 50 mg once daily, 50 mg twice daily or 100 mg once daily. More than 5000 male and more than 4000 female patients received vildagliptin 50 mg once daily or 100 mg daily. More than 1900 patients receiving vildagliptin 50 mg once daily or 100 mg daily were ≥65 years. In these trials, vildagliptin was administered as monotherapy in drug-naïve patients with type 2 diabetes or in combination in patients not adequately controlled by other antidiabetic medicinal products.
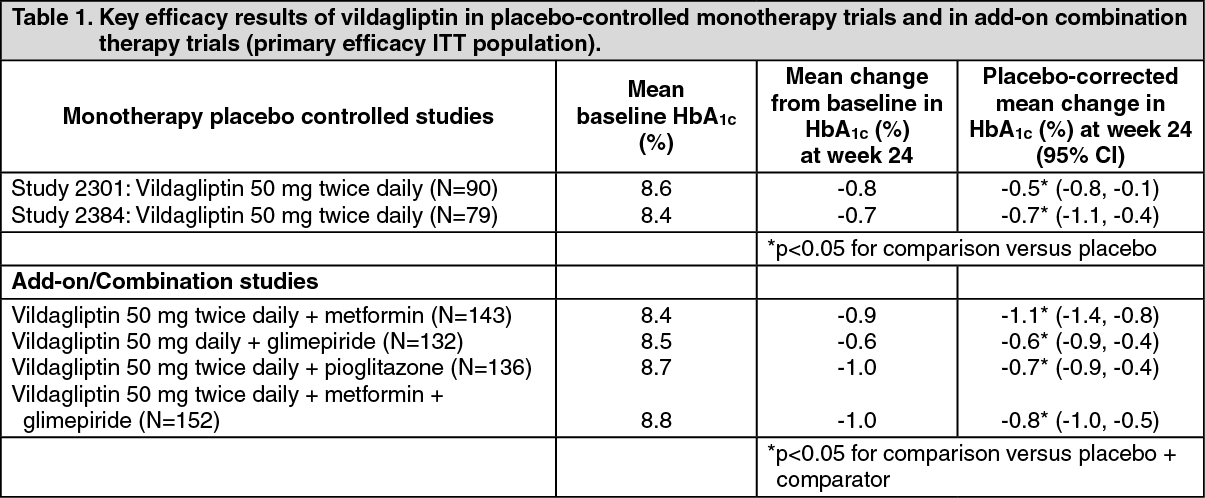
Overall, vildagliptin improved glycaemic control when given as monotherapy or when used in combination with metformin, a sulphonylurea, and a thiazolidinedione, as measured by clinically relevant reductions in HbA
1c from baseline at study endpoint (see Table 1).
In clinical trials, the magnitude of HbA
1c reductions with vildagliptin was greater in patients with higher baseline HbA
1c.
In a 52-week double-blind controlled trial, vildagliptin (50 mg twice daily) reduced baseline HbA
1c by -1% compared to -1.6% for metformin (titrated to 2 g/day) statistical non-inferiority was not achieved. Patients treated with vildagliptin reported significantly lower incidences of gastrointestinal adverse reactions versus those treated with metformin.
In a 24-week double-blind controlled trial, vildagliptin (50 mg twice daily) was compared to rosiglitazone (8 mg once daily). Mean reductions were -1.20% with vildagliptin and -1.48% with rosiglitazone in patients with mean baseline HbA
1c of 8.7%. Patients receiving rosiglitazone experienced a mean increase in weight (+1.6 kg) while those receiving vildagliptin experienced no weight gain (-0.3 kg). The incidence of peripheral oedema was lower in the vildagliptin group than in the rosiglitazone group (2.1% vs. 4.1% respectively).
In a clinical trial of 2 years' duration, vildagliptin (50 mg twice daily) was compared to gliclazide (up to 320 mg/day). After two years, mean reduction in HbA
1c was -0.5% for vildagliptin and -0.6% for gliclazide, from a mean baseline HbA
1c of 8.6%. Statistical non-inferiority was not achieved. Vildagliptin was associated with fewer hypoglycaemic events (0.7%) than gliclazide (1.7%).
In a 24-week trial, vildagliptin (50 mg twice daily) was compared to pioglitazone (30 mg once daily) in patients inadequately controlled with metformin (mean daily dose: 2020 mg). Mean reductions from baseline HbA
1c of 8.4% were -0.9% with vildagliptin added to metformin and -1.0% with pioglitazone added to metformin. A mean weight gain of +1.9 kg was observed in patients receiving pioglitazone added to metformin compared to +0.3 kg in those receiving vildagliptin added to metformin.
In a clinical trial of 2 years' duration, vildagliptin (50 mg twice daily) was compared to glimepiride (up to 6 mg/day - mean dose at 2 years: 4.6 mg) in patients treated with metformin (mean daily dose: 1894 mg). After 1 year mean reductions in HbA
1c were -0.4% with vildagliptin added to metformin and -0.5% with glimepiride added to metformin, from a mean baseline HbA
1c of 7.3%. Body weight change with vildagliptin was -0.2 kg vs +1.6 kg with glimepiride. The incidence of hypoglycaemia was significantly lower in the vildagliptin group (1.7%) than in the glimepiride group (16.2%). At study endpoint (2 years), the HbA
1c was similar to baseline values in both treatment groups and the body weight changes and hypoglycaemia differences were maintained.
In a 52-week trial, vildagliptin (50 mg twice daily) was compared to gliclazide (mean daily dose: 229.5 mg) in patients inadequately controlled with metformin (metformin dose at baseline 1928 mg/day). After 1 year, mean reductions in HbA
1c were -0.81% with vildagliptin added to metformin (mean baseline HbA
1c 8.4%) and -0.85% with gliclazide added to metformin (mean baseline HbA
1c 8.5%); statistical non-inferiority was achieved (95% CI -0.11 - 0.20). Body weight change with vildagliptin was +0.1 kg compared to a weight gain of +1.4 kg with gliclazide.
In a 24-week trial the efficacy of the fixed dose combination of vildagliptin and metformin (gradually titrated to a dose of 50 mg/500 mg twice daily or 50 mg/1000 mg twice daily) as initial therapy in drug-naïve patients was evaluated. Vildagliptin/metformin 50 mg/1000 mg twice daily reduced HbA
1c by -1.82%, vildagliptin/metformin 50 mg/500 mg twice daily by -1.61%, metformin 1000 mg twice daily by -1.36% and vildagliptin 50 mg twice daily by -1.09% from a mean baseline HbA
1c of 8.6%. The decrease in HbA
1c observed in patients with a baseline ≥10.0% was greater.
A 24-week, multi-centre, randomised, double-blind, placebo-controlled trial was conducted to evaluate the treatment effect of vildagliptin 50 mg once daily compared to placebo in 515 patients with type 2 diabetes and moderate renal impairment (N=294) or severe renal impairment (N=221). 68.8% and 80.5% of the patients with moderate and severe renal impairment respectively were treated with insulin (mean daily dose of 56 units and 51.6 units respectively) at baseline. In patients with moderate renal impairment vildagliptin significantly decreased HbA
1c compared with placebo (difference of -0.53%) from a mean baseline of 7.9%. In patients with severe renal impairment, vildagliptin significantly decreased HbA
1c compared with placebo (difference of -0.56%) from a mean baseline of 7.7%.
A 24-week randomised, double-blind, placebo-controlled trial was conducted in 318 patients to evaluate the efficacy and safety of vildagliptin (50 mg twice daily) in combination with metformin (≥1500 mg daily) and glimepiride (≥4 mg daily). Vildagliptin in combination with metformin and glimepiride significantly decreased HbA
1c compared with placebo. The placebo-adjusted mean reduction from a mean baseline HbA
1c of 8.8% was -0.76%.
A 24-week randomised, double-blind, placebo-controlled trial was conducted in 449 patients to evaluate the efficacy and safety of vildagliptin (50 mg twice daily) in combination with a stable dose of basal or premixed insulin (mean daily dose 41 units), with concomitant use of metformin (N=276) or without concomitant metformin (N=173). Vildagliptin in combination with insulin significantly decreased HbA
1c compared with placebo. In the overall population, the placebo-adjusted mean reduction from a mean baseline HbA
1c 8.8% was -0.72%. In the subgroups treated with insulin with or without concomitant metformin the placebo-adjusted mean reduction in HbA
1c was -0.63% and -0.84%, respectively. The incidence of hypoglycaemia in the overall population was 8.4% and 7.2% in the vildagliptin and placebo groups, respectively. Patients receiving vildagliptin experienced no weight gain (+0.2 kg) while those receiving placebo experienced weight reduction (-0.7 kg).
In another 24-week study in patients with more advanced type 2 diabetes not adequately controlled on insulin (short and longer acting, average insulin dose 80 IU/day), the mean reduction in HbA
1c when vildagliptin (50 mg twice daily) was added to insulin was statistically significantly greater than with placebo plus insulin (0.5% vs. 0.2%). The incidence of hypoglycaemia was lower in the vildagliptin group than in the placebo group (22.9% vs. 29.6%).
A 52-week multi-centre, randomised, double-blind trial was conducted in patients with type 2 diabetes and congestive heart failure (NYHA functional class I-III) to evaluate the effect of vildagliptin 50 mg twice daily (N=128) compared to placebo (N=126) on left-ventricular ejection fraction (LVEF). Vildagliptin was not associated with a change in left-ventricular function or worsening of pre-existing CHF. Adjudicated cardiovascular events were balanced overall. There were more cardiac events in vildagliptin-treated patients with NYHA class III heart failure compared to placebo. However, there were imbalances in baseline cardiovascular risk favouring placebo and the number of events was low, precluding firm conclusions. Vildagliptin significantly decreased HbA
1c compared with placebo (difference of 0.6%) from a mean baseline of 7.8% at week 16. In the subgroup with NYHA class III, the decrease in HbA
1c compared to placebo was lower (difference 0.3%) but this conclusion is limited by the small number of patients (n=44). The incidence of hypoglycaemia in the overall population was 4.7% and 5.6% in the vildagliptin and placebo groups, respectively.
A five-year multi-centre, randomised, double-blind study (VERIFY) was conducted in patients with type 2 diabetes to evaluate the effect of an early combination therapy with vildagliptin and metformin (N=998) against standard-of-care initial metformin monotherapy followed by combination with vildagliptin (sequential treatment group) (N=1003) in newly diagnosed patients with type 2 diabetes. The combination regimen of vildagliptin 50 mg twice daily plus metformin resulted in a statistically and clinically significant relative reduction in hazard for "time to confirmed initial treatment failure" (HbA
1c value ≥7%) vs metformin monotherapy in treatment-naïve patients with type 2 diabetes over the 5-year study duration (HR [95% CI]: 0.51 [0.45, 0.58]; p<0.001). The incidence of initial treatment failure (HbA
1c value ≥7%) was 429 (43.6%) patients in the combination treatment group and 614 (62.1%) patients in the sequential treatment group.
Consistently lower HbA
1c was observed with the combination treatment group compared with the sequential treatment group throughout the study duration. An early combination treatment approach with vildagliptin plus metformin in patients with newly diagnosed type 2 diabetes significantly and consistently improved long-term glycaemic durability compared with sequential treatment. The incidence of adverse events (AE) was comparable between the treatment groups (83.5% in the early combination therapy group vs. 83.2% in the sequential treatment group, respectively). The proportion of newly diagnosed patients who experienced hypoglycemic events over the entire study was low in both treatment groups (1.1% in early combination group and 0.6% in sequential treatment group). Both the treatment groups reported microvascular or macrovascular complications in a comparable proportion of patients (30.5% of patients in the early combination group, and 33.1% of patients in the sequential treatment group). The overall safety and tolerability profile was similar between treatment approaches, with no unexpected safety findings reported.
Cardiovascular risk: A meta-analysis of independently and prospectively adjudicated cardiovascular events from 37 phase III and IV monotherapy and combination therapy clinical studies of up to more than 2 years duration (mean exposure 50 weeks for vildagliptin and 49 weeks for comparators) was performed and showed that vildagliptin treatment was not associated with an increase in cardiovascular risk versus comparators. The composite endpoint of adjudicated major adverse cardiovascular events (MACE) including acute myocardial infarction, stroke or cardiovascular death was similar for vildagliptin versus combined active and placebo comparators [Mantel-Haenszel risk ratio (M-H RR) 0.82 (95% CI 0.61-1.11)]. A MACE occurred in 83 out of 9599 (0.86%) vildagliptin-treated patients and in 85 out of 7102 (1.20%) comparator-treated patients. Assessment of each individual MACE component showed no increased risk (similar M-H RR). Confirmed heart failure (HF) events defined as HF requiring hospitalisation or new onset of HF were reported in 41 (0.43%) vildagliptin-treated patients and 32 (0.45%) comparator-treated patients with M-H RR 1.08 (95% CI 0.68-1.70). (See Table 1.)
 Click on icon to see table/diagram/image
Pharmacokinetics: Absorption:
Click on icon to see table/diagram/image
Pharmacokinetics: Absorption: Following oral administration in the fasting state, vildagliptin is rapidly absorbed, with peak plasma concentrations observed at 1.7 hours. Food slightly delays the time to peak plasma concentration to 2.5 hours, but does not alter the overall exposure (AUC). Administration of vildagliptin with food resulted in a decreased C
max (19%). However, the magnitude of change is not clinically significant, so that Galvus can be given with or without food. The absolute bioavailability is 85%.
Distribution: The plasma protein binding of vildagliptin is low (9.3%) and vildagliptin distributes equally between plasma and red blood cells. The mean volume of distribution of vildagliptin at steady-state after intravenous administration (V
ss) is 71 litres, suggesting extravascular distribution.
Biotransformation: Metabolism is the major elimination pathway for vildagliptin in humans, accounting for 69% of the dose. The major metabolite (LAY 151) is pharmacologically inactive and is the hydrolysis product of the cyano moiety, accounting for 57% of the dose, followed by the glucuronide (BQS867) and the amide hydrolysis products (4% of dose).
In vitro data in human kidney microsomes suggest that the kidney may be one of the major organs contributing to the hydrolysis of vildagliptin to its major inactive metabolite, LAY151. DPP-4 contributes partially to the hydrolysis of vildagliptin based on an
in vivo study using DPP-4 deficient rats. Vildagliptin is not metabolised by CYP 450 enzymes to any quantifiable extent. Accordingly, the metabolic clearance of vildagliptin is not anticipated to be affected by co-medications that are CYP 450 inhibitors and/or inducers.
In vitro studies demonstrated that vildagliptin does not inhibit/induce CYP 450 enzymes. Therefore, vildagliptin is not likely to affect metabolic clearance of co-medications metabolised by CYP 1A2, CYP 2C8, CYP 2C9, CYP 2C19, CYP 2D6, CYP 2E1 or CYP 3A4/5.
Elimination: Following oral administration of [
14C] vildagliptin, approximately 85% of the dose was excreted into the urine and 15% of the dose is recovered in the faeces. Renal excretion of the unchanged vildagliptin accounted for 23% of the dose after oral administration. After intravenous administration to healthy subjects, the total plasma and renal clearances of vildagliptin are 41 and 13 l/h, respectively. The mean elimination half-life after intravenous administration is approximately 2 hours. The elimination half-life after oral administration is approximately 3 hours.
Linearity/non-linearity: The C
max for vildagliptin and the area under the plasma concentrations versus time curves (AUC) increased in an approximately dose proportional manner over the therapeutic dose range.
Characteristics in specific groups of patients: Gender: No clinically relevant differences in the pharmacokinetics of vildagliptin were observed between male and female healthy subjects within a wide range of age and body mass index (BMI). DPP-4 inhibition by vildagliptin is not affected by gender.
Elderly: In healthy elderly subjects (≥70 years), the overall exposure of vildagliptin (100 mg once daily) was increased by 32%, with an 18% increase in peak plasma concentration as compared to young healthy subjects (18-40 years). These changes are, however, not considered to be clinically relevant. DPP-4 inhibition by vildagliptin is not affected by age.
Hepatic impairment: The effect of impaired hepatic function on the pharmacokinetics of vildagliptin was studied in patients with mild, moderate and severe hepatic impairment based on the Child-Pugh scores (ranging from 6 for mild to 12 for severe) in comparison with healthy subjects. The exposure to vildagliptin after a single dose in patients with mild and moderate hepatic impairment was decreased (20% and 8%, respectively), while the exposure to vildagliptin for patients with severe impairment was increased by 22%. The maximum change (increase or decrease) in the exposure to vildagliptin is ~30%, which is not considered to be clinically relevant. There was no correlation between the severity of the hepatic disease and changes in the exposure to vildagliptin.
Renal impairment: A multiple-dose, open-label trial was conducted to evaluate the pharmacokinetics of the lower therapeutic dose of vildagliptin (50 mg once daily) in patients with varying degrees of chronic renal impairment defined by creatinine clearance (mild: 50 to <80 ml/min, moderate: 30 to <50 ml/min and severe: <30 ml/min) compared to normal healthy control subjects.
Vildagliptin AUC increased on average 1.4, 1.7 and 2-fold in patients with mild, moderate and severe renal impairment, respectively, compared to normal healthy subjects. AUC of the metabolites LAY151 and BQS867 increased on average about 1.5, 3 and 7-fold in patients with mild, moderate and severe renal impairment, respectively. Limited data from patients with end stage renal disease (ESRD) indicate that vildagliptin exposure is similar to that in patients with severe renal impairment.
LAY151 concentrations were approximately 2-3-fold higher than in patients with severe renal impairment.
Vildagliptin was removed by haemodialysis to a limited extent (3% over a 3-4 hour haemodialysis session starting 4 hours post dose).
Ethnic group: Limited data suggest that race does not have any major influence on vildagliptin pharmacokinetics.
Toxicology: Preclinical safety data: Intra-cardiac impulse conduction delays were observed in dogs with a no-effect dose of 15 mg/kg (7-fold human exposure based on C
max).
Accumulation of foamy alveolar macrophages in the lung was observed in rats and mice. The no-effect dose in rats was 25 mg/kg (5-fold human exposure based on AUC) and in mice 750 mg/kg (142-fold human exposure).
Gastrointestinal symptoms, particularly soft faeces, mucoid faeces, diarrhoea and, at higher doses, faecal blood were observed in dogs. A no-effect level was not established.
Vildagliptin was not mutagenic in conventional
in vitro and
in vivo tests for genotoxicity.
A fertility and early embryonic development study in rats revealed no evidence of impaired fertility, reproductive performance or early embryonic development due to vildagliptin. Embryo-foetal toxicity was evaluated in rats and rabbits. An increased incidence of wavy ribs was observed in rats in association with reduced maternal body weight parameters, with a no-effect dose of 75 mg/kg (10-fold human exposure). In rabbits, decreased foetal weight and skeletal variations indicative of developmental delays were noted only in the presence of severe maternal toxicity, with a no-effect dose of 50 mg/kg (9-fold human exposure). A pre- and postnatal development study was performed in rats. Findings were only observed in association with maternal toxicity at ≥150 mg/kg and included a transient decrease in body weight and reduced motor activity in the F1 generation.
A two-year carcinogenicity study was conducted in rats at oral doses up to 900 mg/kg (approximately 200 times human exposure at the maximum recommended dose). No increases in tumour incidence attributable to vildagliptin were observed. Another two-year carcinogenicity study was conducted in mice at oral doses up to 1000 mg/kg. An increased incidence of mammary adenocarcinomas and haemangiosarcomas was observed with a no-effect dose of 500 mg/kg (59-fold human exposure) and 100 mg/kg (16-fold human exposure), respectively. The increased incidence of these tumours in mice is considered not to represent a significant risk to humans based on the lack of genotoxicity of vildagliptin and its principal metabolite, the occurrence of tumours only in one species and the high systemic exposure ratios at which tumours were observed.
In a 13-week toxicology study in cynomolgus monkeys, skin lesions have been recorded at doses ≥5 mg/kg/day. These were consistently located on the extremities (hands, feet, ears and tail). At 5 mg/kg/day (approximately equivalent to human AUC exposure at the 100 mg dose), only blisters were observed. They were reversible despite continued treatment and were not associated with histopathological abnormalities. Flaking skin, peeling skin, scabs and tail sores with correlating histopathological changes were noted at doses ≥20 mg/kg/day (approximately 3 times human AUC exposure at the 100 mg dose). Necrotic lesions of the tail were observed at ≥80 mg/kg/day. Skin lesions were not reversible in the monkeys treated at 160 mg/kg/day during a 4-week recovery period.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
