


 Sign Out
Sign Out




Pharmacology: Pharmacodynamics: Mechanism of action: Dapagliflozin is a highly potent (Ki: 0.55 nM), selective and reversible inhibitor of SGLT2.
Inhibition of SGLT2 by dapagliflozin reduces reabsorption of glucose from the glomerular filtrate in the proximal renal tubule with a concomitant reduction in sodium reabsorption leading to urinary excretion of glucose and osmotic diuresis. Dapagliflozin therefore increases the delivery of sodium to the distal tubule which increases tubuloglomerular feedback and reduces intraglomerular pressure. This combined with osmotic diuresis leads to a reduction in volume overload, reduced blood pressure, and lower preload and afterload, which may have beneficial effects on cardiac remodelling and diastolic function, and preserve renal function. The cardiac and renal benefits of dapagliflozin are not solely dependent on the blood glucose-lowering effect and not limited to patients with diabetes as demonstrated in the DAPA-HF, DELIVER and DAPA-CKD studies. Other effects include an increase in haematocrit and reduction in body weight.
Dapagliflozin improves both fasting and post-prandial plasma glucose levels by reducing renal glucose reabsorption leading to urinary glucose excretion. This glucose excretion (glucuretic effect) is observed after the first dose, is continuous over the 24-hour dosing interval and is sustained for the duration of treatment. The amount of glucose removed by the kidney through this mechanism is dependent upon the blood glucose concentration and GFR. Thus, in subjects with normal blood glucose, dapagliflozin has a low propensity to cause hypoglycaemia. Dapagliflozin does not impair normal endogenous glucose production in response to hypoglycaemia. Dapagliflozin acts independently of insulin secretion and insulin action. Improvement in homeostasis model assessment for beta cell function (HOMA beta-cell) has been observed in clinical studies with dapagliflozin.
The SGLT2 is selectively expressed in the kidney. Dapagliflozin does not inhibit other glucose transporters important for glucose transport into peripheral tissues and is >1,400 times more selective for SGLT2 versus SGLT1, the major transporter in the gut responsible for glucose absorption.
Pharmacodynamic effects: Increases in the amount of glucose excreted in the urine were observed in healthy subjects and in subjects with type 2 diabetes mellitus following the administration of dapagliflozin. Approximately 70 g of glucose was excreted in the urine per day (corresponding to 280 kcal/day) at a dapagliflozin dose of 10 mg/day in subjects with type 2 diabetes mellitus for 12 weeks. Evidence of sustained glucose excretion was seen in subjects with type 2 diabetes mellitus given dapagliflozin 10 mg/day for up to 2 years.
This urinary glucose excretion with dapagliflozin also results in osmotic diuresis and increases in urinary volume in subjects with type 2 diabetes mellitus. Urinary volume increases in subjects with type 2 diabetes mellitus treated with dapagliflozin 10 mg were sustained at 12 weeks and amounted to approximately 375 mL/day. The increase in urinary volume was associated with a small and transient increase in urinary sodium excretion that was not associated with changes in serum sodium concentrations.
Urinary uric acid excretion was also increased transiently (for 3-7 days) and accompanied by a sustained reduction in serum uric acid concentration. At 24 weeks, reductions in serum uric acid concentrations ranged from -48.3 to -18.3 micromoles/L (-0.87 to -0.33 mg/dL).
Clinical efficacy and safety: Type 2 diabetes mellitus: Improvement of glycemic control and reduction of cardiovascular and renal morbidity and mortality are integral parts of the treatment of type 2 diabetes.
Fourteen double-blind, randomised, controlled clinical trials were conducted with 7,056 subjects with type 2 diabetes to evaluate the glycaemic efficacy and safety of Forxiga; 4,737 subjects in these studies were treated with dapagliflozin. Twelve studies had a treatment period of 24 weeks duration, 8 with long-term extensions ranging from 24 to 80 weeks (up to a total study duration of 104 weeks), and one study had a 28-week treatment period, and one study was 52 weeks in duration with long-term extensions of 52 and 104 weeks (total study duration of 208 weeks). Mean duration of diabetes ranged from 1.4 to 16.9 years. Fifty percent (50%) had mild renal impairment and 11% had moderate renal impairment. Fifty-one percent (51%) of the subjects were men, 84% were White, 8% were Asian, 4% were Black and 4% were of other racial groups. Eighty-one percent (81%) of the subjects had a body mass index (BMI) ≥ 27. Furthermore, two 12-week, placebo-controlled studies were conducted in patients with inadequately controlled type 2 diabetes and hypertension.
A cardiovascular outcomes study (DECLARE) was conducted with dapagliflozin 10 mg compared with placebo in 17,160 patients with type 2 diabetes mellitus with or without established cardiovascular disease to evaluate the effect on cardiovascular and renal events.
Glycaemic control: Monotherapy: A double-blind, placebo-controlled study of 24-week duration (with an additional extension period) was conducted to evaluate the safety and efficacy of monotherapy with Forxiga in subjects with inadequately controlled type 2 diabetes mellitus. Once-daily treatment with dapagliflozin resulted in statistically significant (p <0.0001) reductions in HbA1c compared to placebo (Table 1).
In the extension period, HbA1c reductions were sustained through week 102 (-0.61%, and -0.17% adjusted mean change from baseline for dapagliflozin 10 mg and placebo, respectively). (See Table 1.)
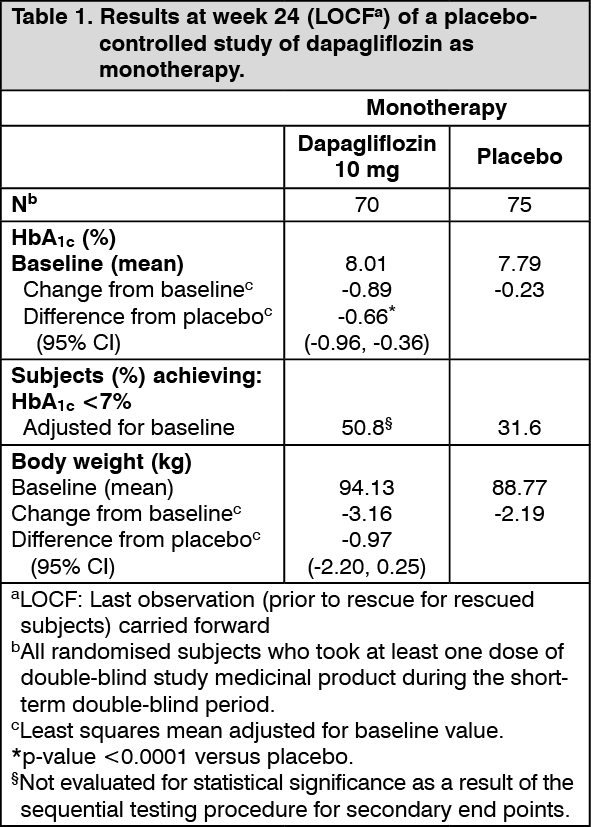 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdd-on combination therapy: In a 52-week, active-controlled non-inferiority study (with 52- and 104-week extension periods), Forxiga was evaluated as add-on therapy to metformin compared with a sulphonylurea (glipizide) as add-on therapy to metformin in subjects with inadequate glycaemic control (HbA1c >6.5% and ≤10%). The results showed a similar mean reduction in HbA1c from baseline to week 52, compared to glipizide, thus demonstrating non-inferiority (Table 2). At week 104, adjusted mean change from baseline in HbA1c was -0.32% for dapagliflozin and -0.14% for glipizide. At week 208, adjusted mean change from baseline in HbA1c was -0.10% for dapagliflozin and 0.20% for glipizide. At 52, 104 and 208 weeks, a significantly lower proportion of subjects in the group treated with dapagliflozin (3.5%, 4.3% and 5.0%, respectively) experienced at least one event of hypoglycaemia compared to the group treated with glipizide (40.8%, 47.0% and 50.0%, respectively). The proportion of subjects remaining in the study at week 104 and week 208 was 56.2% and 39.7% for the group treated with dapagliflozin and 50.0% and 34.6% for the group treated with glipizide. (See Table 2.)
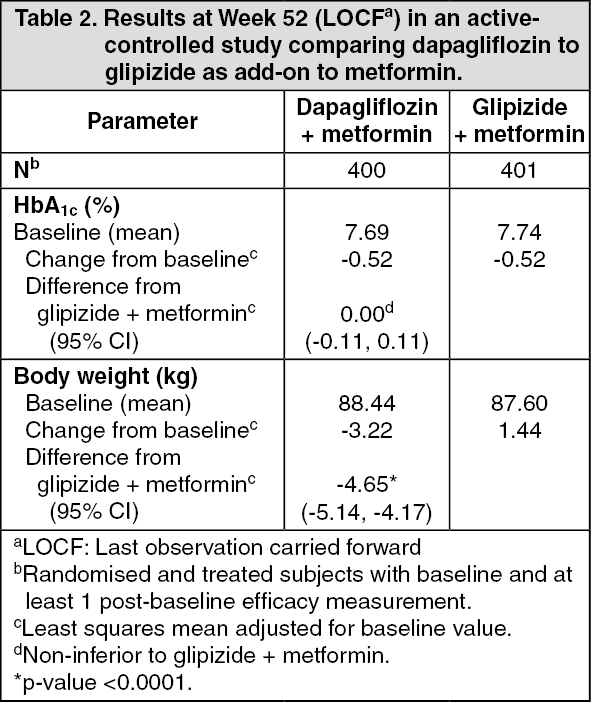 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDapagliflozin as an add-on with either metformin, glimepiride, metformin and a sulphonylurea, sitagliptin (with or without metformin) or insulin resulted in statistically significant reductions in HbA1c at 24 weeks compared with subjects receiving placebo (p <0.0001; Tables 3, 4 and 5).
The reductions in HbA1c observed at week 24 were sustained in add-on combination studies (glimepiride and insulin) with 48-week data (glimepiride) and up to 104-week data (insulin). At week 48 when added to sitagliptin (with or without metformin), the adjusted mean change from baseline for dapagliflozin 10 mg and placebo was -0.30% and 0.38%, respectively. For the add-on to metformin study, HbA1c reductions were sustained through week 102 (-0.78% and 0.02% adjusted mean change from baseline for 10 mg and placebo, respectively). At week 104 for insulin (with or without additional oral glucose-lowering medicinal products), the HbA1c reductions were -0.71% and -0.06% adjusted mean change from baseline for dapagliflozin 10 mg and placebo, respectively. At weeks 48 and 104, the insulin dose remained stable compared to baseline in subjects treated with dapagliflozin 10 mg at an average dose of 76 IU/day. In the placebo group there was a mean increase of 10.5 IU/day and 18.3 IU/day from baseline (mean average dose of 84 and 92 IU/day) at weeks 48 and 104, respectively. The proportion of subjects remaining in the study at week 104 was 72.4% for the group treated with dapagliflozin 10 mg and 54.8% for the placebo group. (See Tables 3, 4 and 5.)
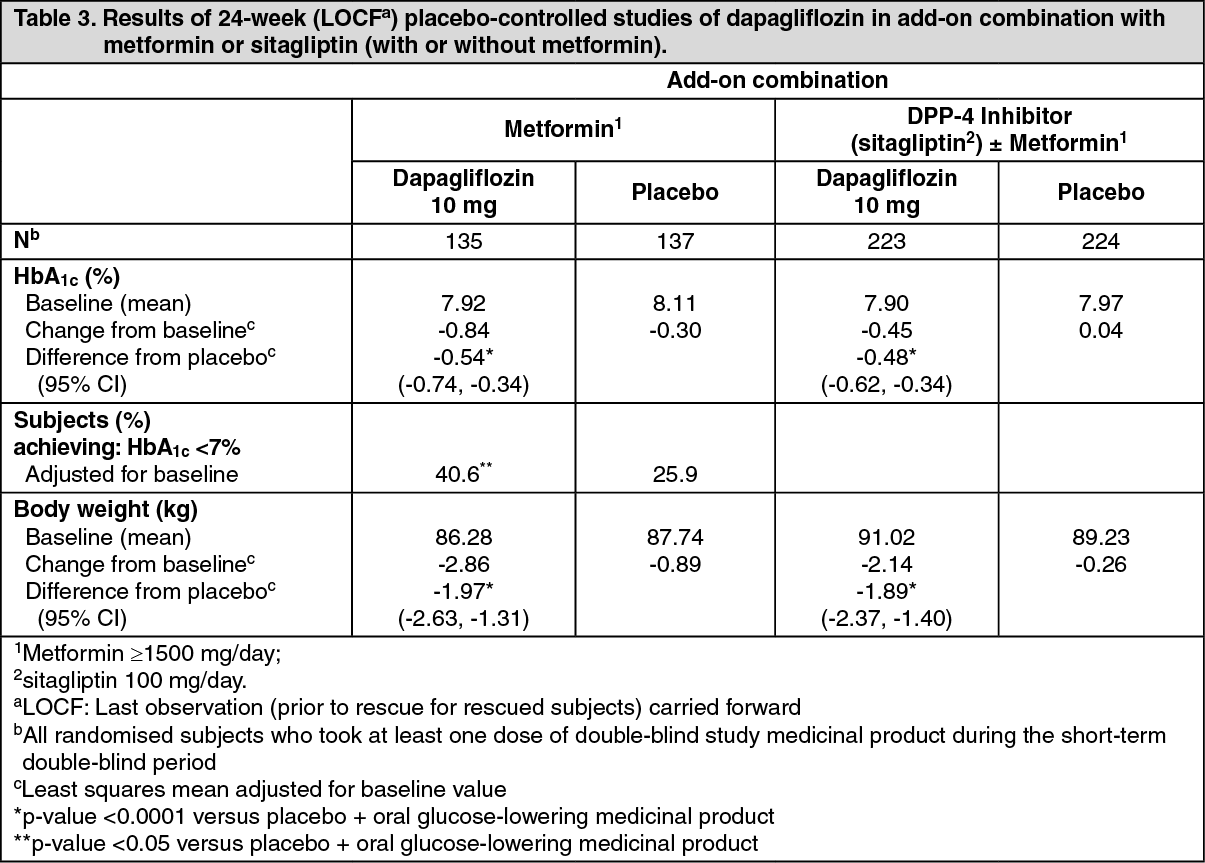 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image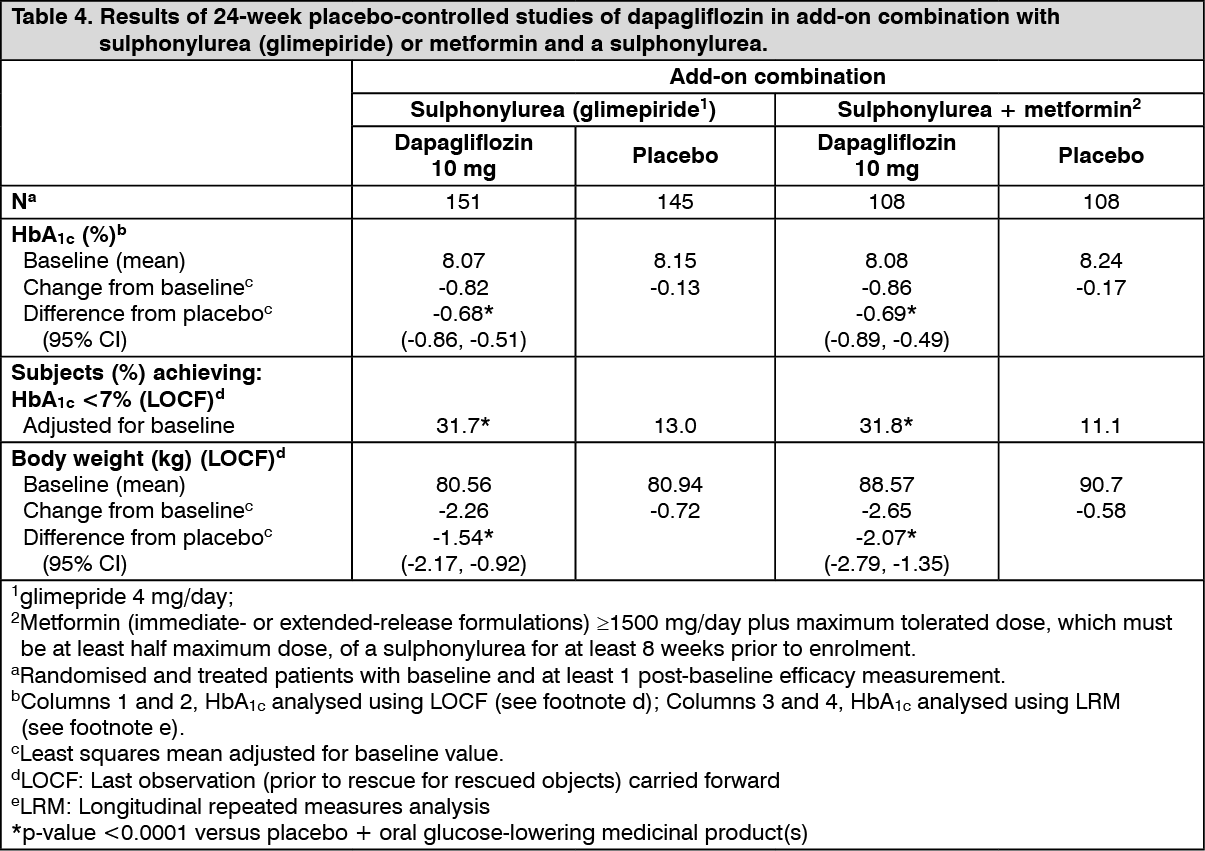 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image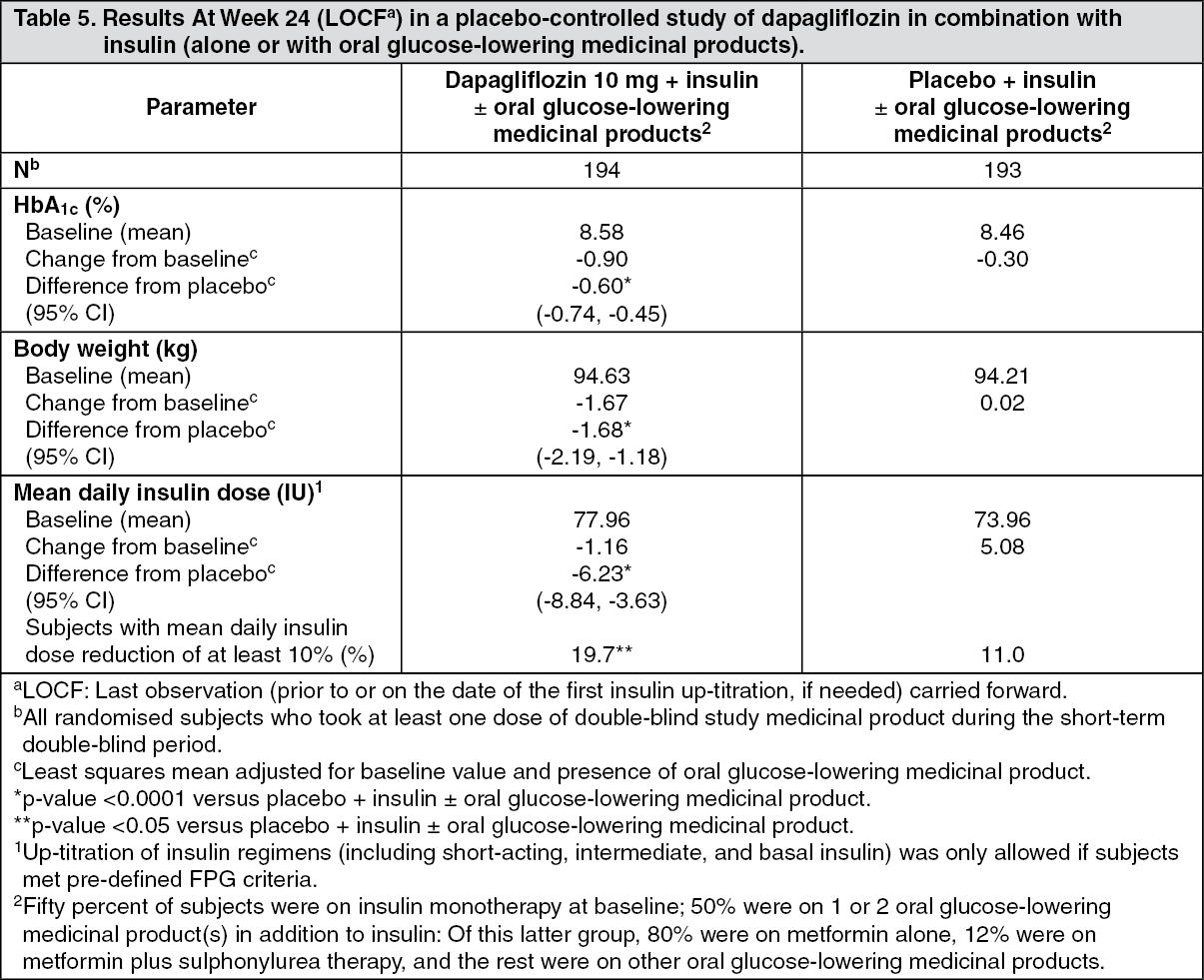 Click on icon to see table/diagram/image
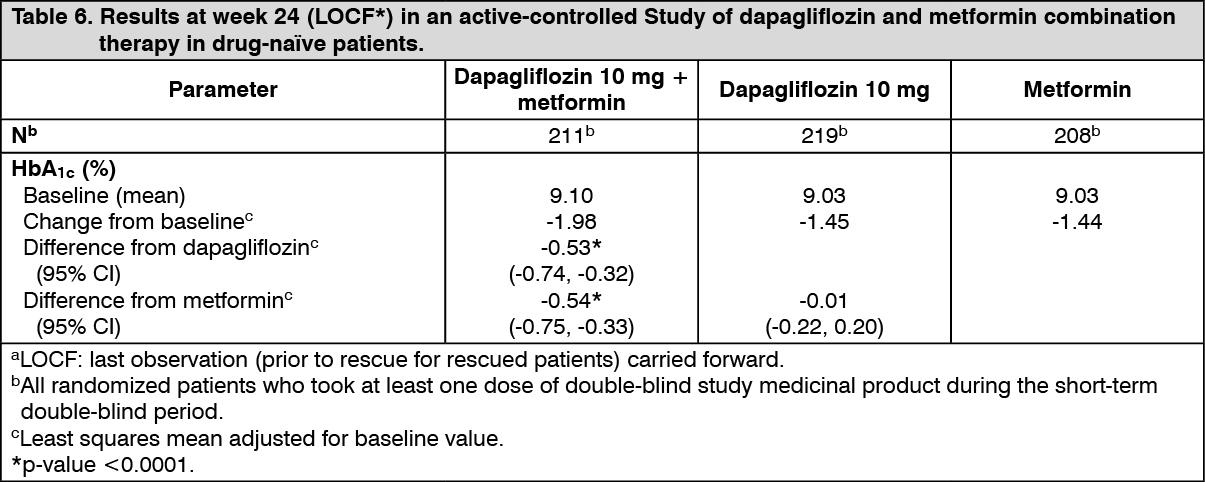
Click on icon to see table/diagram/imageIn combination with metformin in drug-naïve patients: A total of 1,236 drug-naive patients with inadequately controlled type 2 diabetes (HbA1c ≥ 7.5% and ≤ 12%) participated in two active-controlled studies of 24 weeks duration to evaluate the efficacy and safety of dapagliflozin (5 mg or 10 mg) in combination with metformin in drug-naïve patients versus therapy with the monocomponents.
Treatment with dapagliflozin 10 mg in combination with metformin (up to 2000 mg per day) provided significant improvements in HbA1c compared to the individual components (Table 6), and led to greater reductions in fasting plasma glucose (FPG) (compared to the individual components) and body weight (compared to metformin). (See Table 6.)
 Click on icon to see table/diagram/image
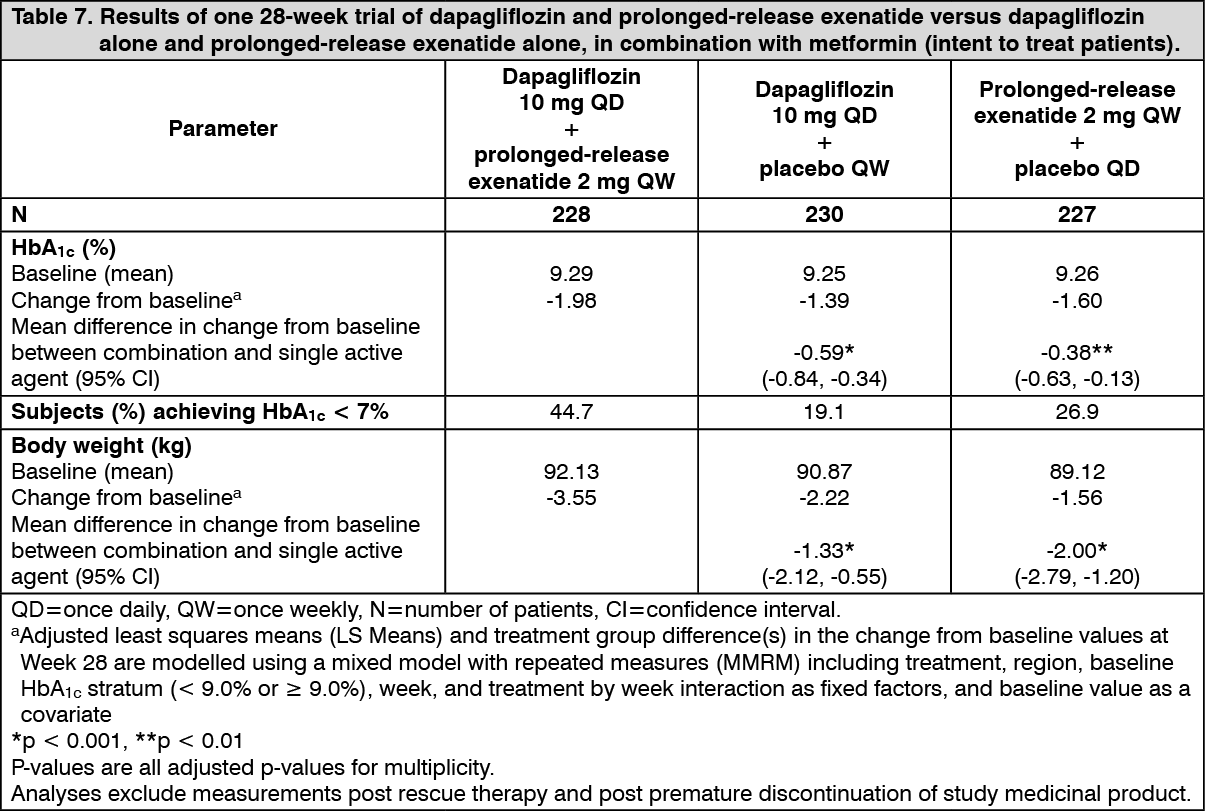
Click on icon to see table/diagram/imageCombination therapy with prolonged-release exenatide: In a 28-week, double-blind, active comparator-controlled study, the combination of dapagliflozin and prolonged-release exenatide (a GLP-1 receptor agonist) was compared to dapagliflozin alone and prolonged-release exenatide alone in subjects with inadequate glycaemic control on metformin alone (HbA1c ≥ 8% and ≤ 12%). All treatment groups had a reduction in HbA1c compared to baseline. The combination treatment with dapagliflozin 10 mg and prolonged-release exenatide group showed superior reductions in HbA1c from baseline compared to dapagliflozin alone and prolonged-release exenatide alone (Table 7). (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFasting plasma glucose: Treatment with dapagliflozin 10 mg as a monotherapy or as an add-on to either metformin, glimepiride, metformin and a sulphonylurea, sitagliptin (with or without metformin) or insulin resulted in statistically significant reductions in FPG (-1.90 to -1.20 mmol/L [-34.2 to -21.7 mg/dL]) compared to placebo (-0.33 to 0.21 mmol/L [-6.0 to 3.8 mg/dL]). This effect was observed at week 1 of treatment and maintained in studies extended through week 104.
Combination therapy of dapagliflozin 10 mg and prolonged-release exenatide resulted in significantly greater reductions in FPG at week 28: -3.66 mmol/l (-65.8 mg/dl), compared to -2.73 mmol/l (-49.2 mg/dl) for dapagliflozin alone (p <0.001) and -2.54 mmol/l (-45.8 mg/dl) for exenatide alone (p <0.001).
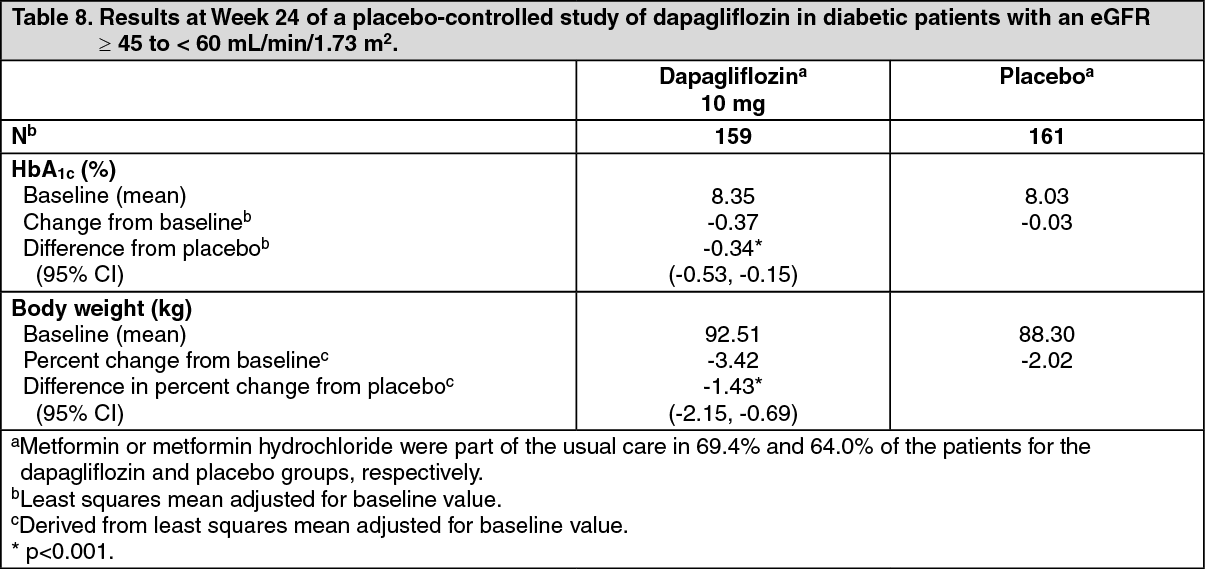
In a dedicated study in diabetic patients with an eGFR ≥ 45 to <60 mL/min/1.73 m2, treatment with dapagliflozin demonstrated reductions in FPG at week 24: -1.19 mmol/L (-21.46 mg/dL) compared to -0.27 mmol/L (-4.87 mg/dL) for placebo (p=0.001).
Post-prandial glucose: Treatment with dapagliflozin 10 mg as an add-on to glimepiride resulted in statistically significant reductions in 2-hour post-prandial glucose at 24 weeks that were maintained up to week 48.
Treatment with dapagliflozin 10 mg as an add-on to sitagliptin (with or without metformin) resulted in reductions in 2-hour post-prandial glucose at 24 weeks that were maintained up to week 48.
Combination therapy of dapagliflozin 10 mg and prolonged-release exenatide resulted in significantly greater reductions in 2-hour post-prandial glucose at week 28 compared to either medicinal product alone.
Body weight: Dapagliflozin 10 mg as an add-on to metformin, glimepiride, metformin and a sulphonylurea, sitagliptin (with or without metformin) or insulin resulted in statistically significant body weight reduction at 24 weeks (p <0.0001, Tables 3 and 4). These effects were sustained in longer-term trials. At 48 weeks, the difference for dapagliflozin as add-on to sitagliptin (with or without metformin) compared with placebo was -2.22 kg. At 102 weeks, the difference for dapagliflozin as add-on to metformin compared with placebo, or as add-on to insulin compared with placebo was -2.14 and -2.88 kg, respectively.
As an add-on therapy to metformin in an active-controlled non-inferiority study, dapagliflozin resulted in a statistically significant body weight reduction compared with glipizide of -4.65 kg at 52 weeks (p <0.0001, Table 2) that was sustained at 104 and 208 weeks (-5.06 kg and -4.38 kg, respectively).
The combination of dapagliflozin 10 mg and prolonged-release exenatide demonstrated significantly greater weight reductions compared to either medicinal product alone (Table 7).
A 24-week study in 182 diabetic subjects using dual energy X-ray absorptiometry (DXA) to evaluate body composition demonstrated reductions with dapagliflozin 10 mg plus metformin compared with placebo plus metformin, respectively, in body weight and body fat mass as measured by DXA rather than lean tissue or fluid loss. Treatment with Forxiga plus metformin showed a numerical decrease in visceral adipose tissue compared with placebo plus metformin treatment in a magnetic resonance imaging substudy.
Blood pressure: In a pre-specified pooled analysis of 13 placebo-controlled studies, treatment with dapagliflozin 10 mg resulted in a systolic blood pressure change from baseline of -3.7 mmHg and diastolic blood pressure of -1.8 mmHg versus -0.5 mmHg systolic and -0.5 mmHg diastolic blood pressure for placebo group at week 24. Similar reductions were observed up to 104 weeks.
Combination therapy of dapagliflozin 10 mg and prolonged-release exenatide resulted in a significantly greater reduction in systolic blood pressure at week 28 (-4.3 mmHg) compared to dapagliflozin alone (-1.8 mmHg, p <0.05) and prolonged-release exenatide alone (-1.2 mmHg, p <0.01).
In two 12-week, placebo-controlled studies a total of 1,062 patients with inadequately controlled type 2 diabetes and hypertension (despite pre-existing stable treatment with an ACE-I or ARB in one study and an ACE-I or ARB plus one additional antihypertensive treatment in another study) were treated with dapagliflozin 10 mg or placebo. At week 12 for both studies, dapagliflozin 10 mg plus usual antidiabetic treatment provided improvement in HbA1c and decreased the placebo-corrected systolic blood pressure on average by 3.1 and 4.3 mmHg, respectively.
In a dedicated study in diabetic patients with an eGFR ≥45 to <60 mL/min/1.73 m2, treatment with dapagliflozin demonstrated reductions in seated systolic blood pressure at week 24: -4.8 mmHg compared to -1.7 mmHg for placebo (p <0.05).
Glycaemic control in patients with moderate renal impairment CKD 3A (eGFR ≥45 to <60 mL/min/1.73 m2): The efficacy of dapagliflozin was assessed in a dedicated study in diabetic patients with an eGFR ≥45 to <60 mL/min/1.73 m2 who had inadequate glycaemic control on usual care. Treatment with dapagliflozin resulted in reductions in HbA1c and body weight compared with placebo (Table 8). (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients with baseline HbA1c ≥9%: In a pre-specified analysis of subjects with baseline HbA1c ≥9.0%, treatment with dapagliflozin 10 mg resulted in statistically significant reductions in HbA1c at week 24 as a monotherapy (adjusted mean change from baseline: -2.04% and 0.19% for dapagliflozin 10 mg and placebo, respectively) and as an add-on to metformin (adjusted mean change from baseline: -1.32% and -0.53% for dapagliflozin and placebo, respectively).
Cardiovascular and renal outcomes: Dapagliflozin Effect on Cardiovascular Events (DECLARE) was an international, multicentre, randomised, double-blind, placebo-controlled clinical study conducted to determine the effect of dapagliflozin compared with placebo on cardiovascular outcomes when added to current background therapy. All patients had type 2 diabetes mellitus and either at least two additional cardiovascular risk factors (age ≥55 years in men or ≥60 years in women and one or more of dyslipidaemia, hypertension or current tobacco use) or established cardiovascular disease.
Of 17,160 randomised patients, 6,974 (40.6%) had established cardiovascular disease and 10,186 (59.4%) did not have established cardiovascular disease. 8,582 patients were randomised to dapagliflozin 10 mg and 8,578 to placebo, and were followed for a median of 4.2 years.
The mean age of the study population was 63.9 years, 37.4% were female. In total, 22.4% had diabetes for ≤5 years, mean duration of diabetes was 11.9 years. Mean HbA1c was 8.3% and mean BMI was 32.1 kg/m2.
At baseline, 10.0% of patients had a history of heart failure. Mean eGFR was 85.2 mL/min/1.73 m2, 7.4% of patients had eGFR <60 mL/min/1.73 m2, and 30.3% of patients had micro- or macroalbuminuria (UACR ≥30 to ≤300 mg/g or >300 mg/g, respectively).
Most patients (98%) used one or more diabetic medicinal products at baseline, including metformin (82%), insulin (41%) and sulfonylurea (43%).
The primary endpoints were time to first event of the composite of cardiovascular death, myocardial infarction or ischaemic stroke (MACE) and time to first event of the composite of hospitalisation for heart failure or cardiovascular death. The secondary endpoints were a renal composite endpoint and all-cause mortality.
Major adverse cardiovascular events: Dapagliflozin 10 mg demonstrated non-inferiority versus placebo for the composite of cardiovascular death, myocardial infarction or ischaemic stroke (one-sided p <0.001).
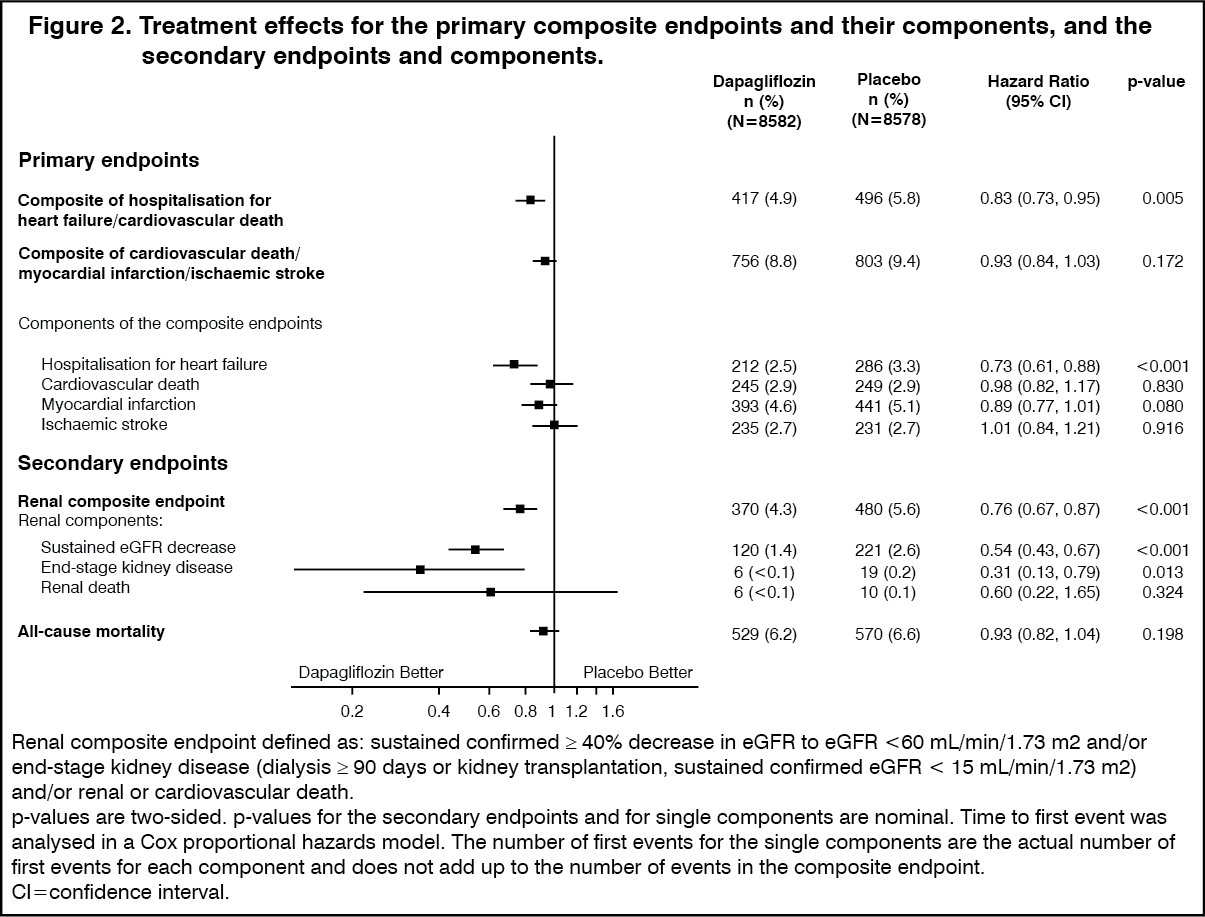
Heart failure or cardiovascular death: Dapagliflozin 10 mg demonstrated superiority versus placebo in preventing the composite of hospitalisation for heart failure or cardiovascular death (Figure 1). The difference in treatment effect was driven by hospitalisation for heart failure, with no difference in cardiovascular death (Figure 2).
The treatment benefit of dapagliflozin over placebo was observed both in patients with and without established cardiovascular disease, with and without heart failure at baseline, and was consistent across key subgroups, including age, gender, renal function (eGFR) and region. (See Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageResults on primary and secondary endpoints are displayed in Figure 2. Superiority of dapagliflozin over placebo was not demonstrated for MACE (p=0.172). The renal composite endpoint and all-cause mortality were therefore not tested as part of the confirmatory testing procedure. (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageNephropathy: Dapagliflozin reduced the incidence of events of the composite of confirmed sustained eGFR decrease, end-stage kidney disease, renal or cardiovascular death. The difference between groups was driven by reductions in events of the renal components; sustained eGFR decrease, end-stage kidney disease and renal death (Figure 2).
The hazard ratio (HR) for time to nephropathy (sustained eGFR decrease, end-stage kidney disease and renal death) was 0.53 (95% CI 0.43, 0.66) for dapagliflozin versus placebo.
In addition, dapagliflozin reduced the new onset of sustained albuminuria (HR 0.79 [95% CI 0.72, 0.87]) and led to greater regression of macroalbuminuria (HR 1.82 [95% CI 1.51, 2.20]) compared with placebo.
Heart failure: DAPA-HF study: Heart failure with reduced ejection fraction (LVEF ≤40%): Dapagliflozin And Prevention of Adverse outcomes in Heart Failure (DAPA-HF) was an international, multicentre, randomised, double-blind, placebo-controlled study in patients with heart failure (New York Heart Association [NYHA] functional class II-IV) with reduced ejection fraction (left ventricular ejection fraction [LVEF] ≤40%) to determine the effect of dapagliflozin compared with placebo, when added to background standard of care therapy, on the incidence of cardiovascular death and worsening heart failure.
Of 4,744 patients, 2,373 were randomised to dapagliflozin 10 mg and 2,371 to placebo and followed for a median of 18 months. The mean age of the study population was 66 years, 77% were male.
At baseline, 67.5% of the patients were classified as NYHA class II, 31.6% class III and 0.9% class IV, median LVEF was 32%, 56% of the heart failures were ischaemic, 36% were non-ischaemic and 8% were of unknown aetiology. In each treatment group, 42% of the patients had a history of type 2 diabetes mellitus, and an additional 3% of the patients in each group were classified as having type 2 diabetes mellitus based on a HbA1c ≥6.5% at both enrolment and randomisation. Patients were on standard of care therapy; 94% of patients were treated with ACE-I, ARB or angiotensin receptor-neprilysin inhibitor (ARNI,11%), 96% with beta-blocker, 71% with mineralocorticoid receptor antagonist (MRA), 93% with diuretic and 26% had an implantable device (with defibrillator function).
Patients with eGFR ≥30 ml/min/1.73 m2 at enrolment were included in the study. The mean eGFR was 66 ml/min/1.73 m2, 41% of patients had eGFR <60 ml/min/1.73 m2 and 15% had eGFR <45 ml/min/1.73 m2.
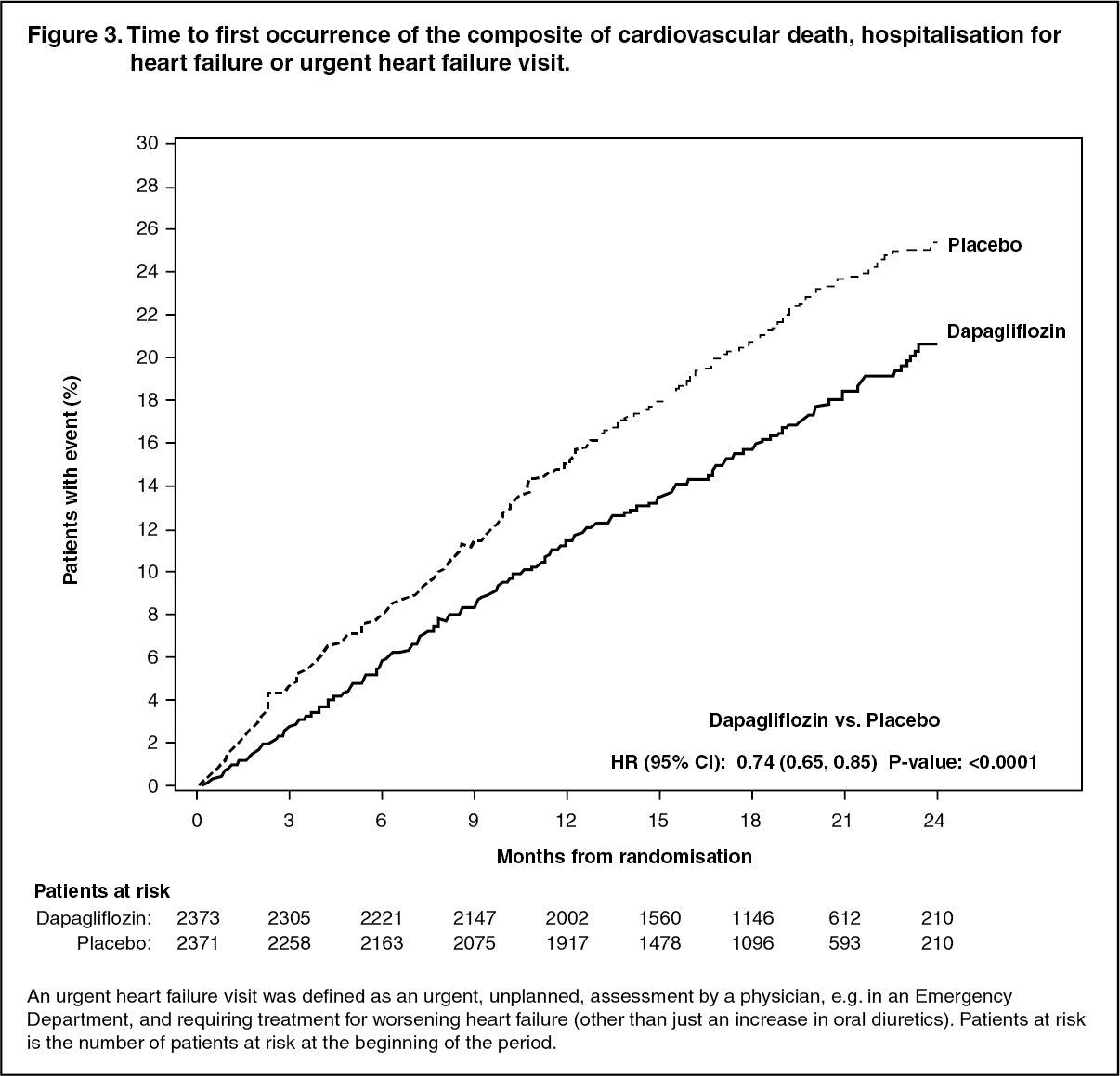
Cardiovascular death and worsening heart failure: Dapagliflozin was superior to placebo in preventing the primary composite endpoint of cardiovascular death, hospitalisation for heart failure or urgent heart failure visit (HR 0.74 [95% CI 0.65, 0.85], p <0.0001). The effect was observed early and was sustained throughout the duration of the study (Figure 3). (See Figure 3.)
 Click on icon to see table/diagram/image
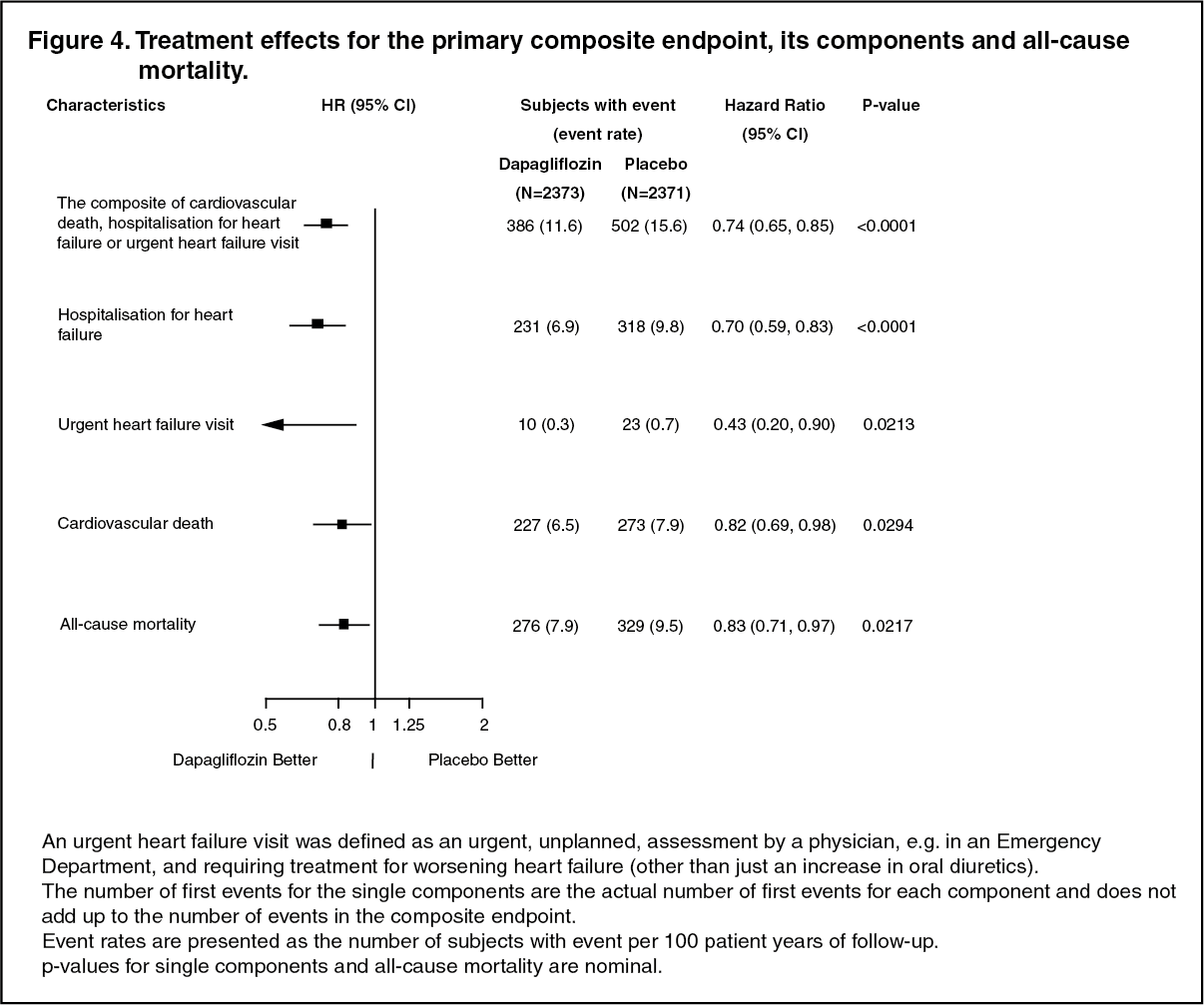
Click on icon to see table/diagram/imageAll three components of the primary composite endpoint individually contributed to the treatment effect (Figure 4). There were few urgent heart failure visits. (See Figure 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDapagliflozin also reduced the total number of events of hospitalisations for heart failure (first and recurrent) and cardiovascular death; there were 567 events in the dapagliflozin group versus 742 events in the placebo group (Rate Ratio 0.75 [95% CI 0.65, 0.88]; p=0.0002).
The treatment benefit of dapagliflozin was observed in heart failure patients both with type 2 diabetes mellitus and without diabetes. Dapagliflozin reduced the primary composite endpoint of incidence of cardiovascular death and worsening heart failure with a HR of 0.75 (95% CI 0.63, 0.90) in patients with diabetes and 0.73 (95% CI 0.60, 0.88) in patients without diabetes.
The treatment benefit of dapagliflozin over placebo on the primary endpoint was also consistent across other key subgroups, including concomitant heart failure therapy, renal function (eGFR), age, gender, and region.
Patient reported outcome - heart failure symptoms: The treatment effect of dapagliflozin on heart failure symptoms was assessed by the Total Symptom Score of the Kansas City Cardiomyopathy Questionnaire (KCCQ-TSS), which quantifies heart failure symptom frequency and severity, including fatigue, peripheral oedema, dyspnoea and orthopnoea. The score ranges from 0 to 100, with higher scores representing better health status.
Treatment with dapagliflozin resulted in a statistically significant and clinically meaningful benefit over placebo in heart failure symptoms, as measured by change from baseline at month 8 in the KCCQ-TSS. (Win Ratio 1.18 [95% CI 1.11, 1.26]; p <0.0001). Both symptom frequency and symptom burden contributed to the results. Benefit was seen both in improving heart failure symptoms and in preventing deterioration of heart failure symptoms.
In responder analyses, the proportion of patients with a clinically meaningful improvement on the KCCQ-TSS from baseline at 8 months, defined as 5 points or more, was higher for the dapagliflozin treatment group compared with placebo. The proportion of patients with a clinically meaningful deterioration, defined as 5 points or more, was lower for the dapagliflozin treatment group compared to placebo. The benefits observed with dapagliflozin remained when applying more conservative cut-offs for larger clinically meaningful change (Table 9). (See Table 9.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageNephropathy: There were few events of the renal composite endpoint (confirmed sustained ≥50% eGFR decrease, ESKD, or renal death); the incidence was 1.2% in the dapagliflozin group and 1.6% in the placebo group.
DELIVER study: Heart failure with left ventricular ejection fraction >40%: Dapagliflozin Evaluation to Improve the LIVEs of Patients with PReserved Ejection Fraction Heart Failure (DELIVER) was an international, multicentre, randomised, double-blind, placebo-controlled study in patients aged ≥40 years with heart failure (NYHA class II-IV) with LVEF > 40% and evidence of structural heart disease, to determine the effect of dapagliflozin compared with placebo on the incidence of cardiovascular death and worsening heart failure.
Of 6,263 patients, 3,131 were randomised to dapagliflozin 10 mg and 3,132 to placebo and followed for a median of 28 months. The study included 654 (10%) subacute heart failure patients (defined as randomised during hospitalisation for heart failure or within 30 days of discharge). The mean age of the study population was 72 years and 56% were male.
At baseline, 75% patients were classified as NYHA class II, 24% class III and 0.3% class IV. Median LVEF was 54%, 34% of the patients had LVEF ≤49%, 36% had LVEF 50-59% and 30% had LVEF ≥60%. In each treatment group, 45% had a history of type 2 diabetes mellitus. Baseline therapy included ACEi/ARB/ARNI (77%), beta-blockers (83%) diuretics (98%) and MRA (43%).
The mean eGFR was 61 mL/min/1.73 m2, 49% of patients had eGFR <60mL/min/1.73 m2, 23% had eGFR <45 mL/min/1.73 m2, and 3% had eGFR <30 mL/min/1.73 m2.
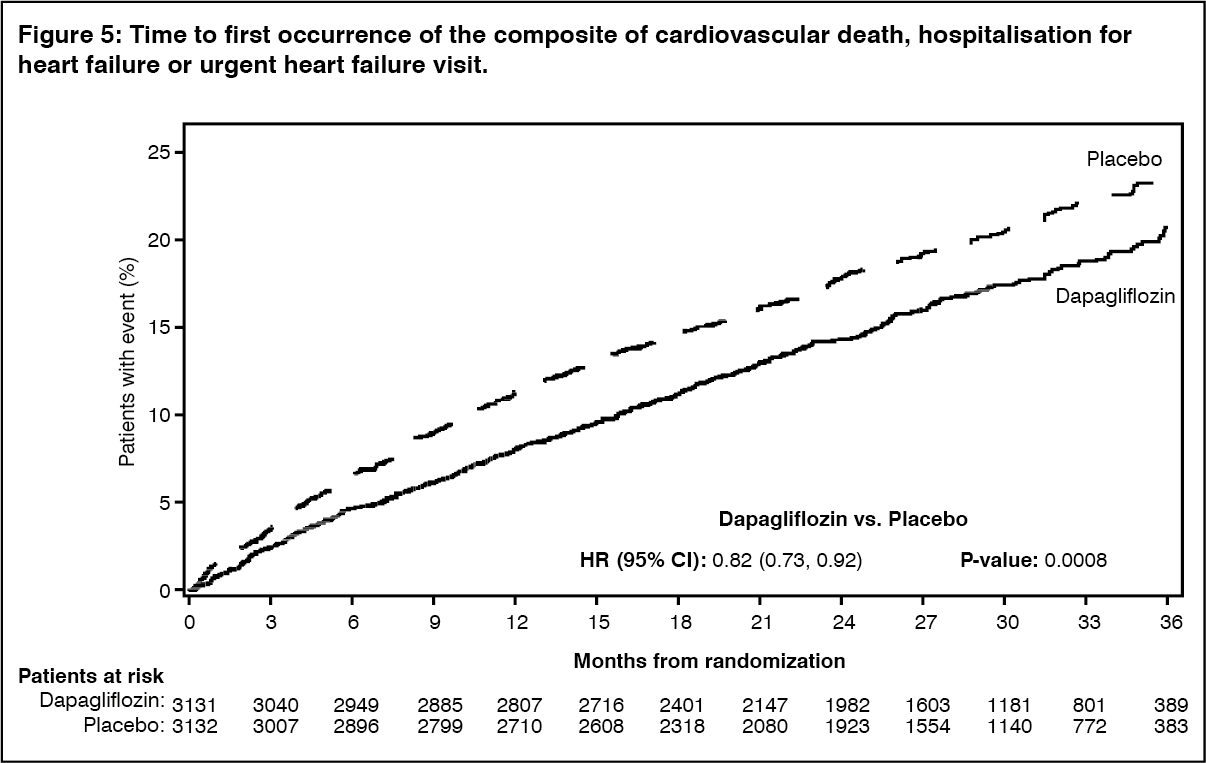
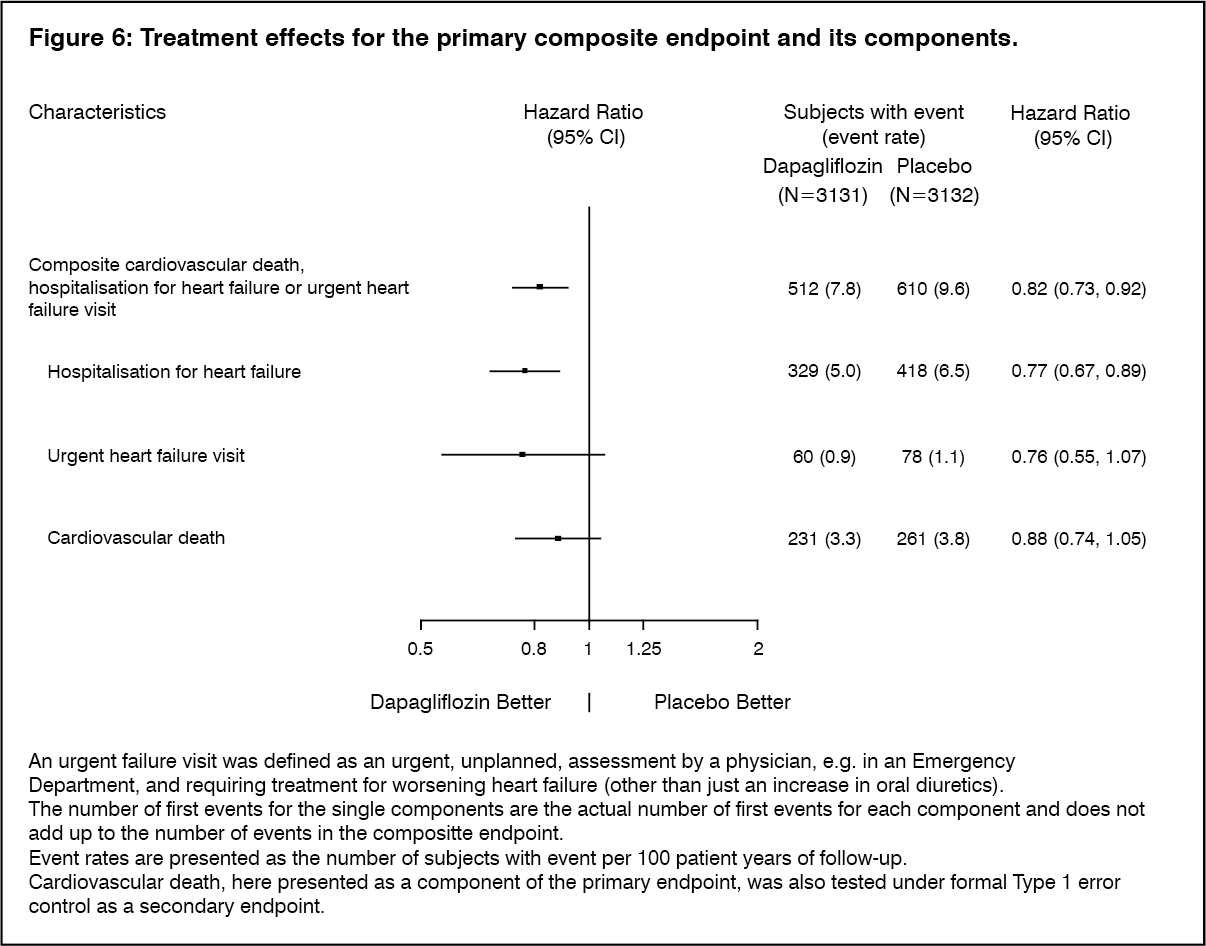
Dapagliflozin was superior to placebo in reducing the incidence of the primary composite endpoint of cardiovascular death, hospitalisation for heart failure or urgent heart failure visit (HR 0.82 [95% CI 0.73, 0.92]; p=0.0008) (Figure 5). (see Figure 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFigure 6 presents the contribution of the three components of the primary composite endpoint to the treament effect. (See Figure 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageChronic kidney disease: The Study to Evaluate the Effect of Dapagliflozin on Renal Outcomes and Cardiovascular Mortality in Patients with Chronic Kidney Disease (DAPA-CKD) was an international, multicentre, randomised, double-blind, placebo-controlled study in patients with chronic kidney disease (CKD) with eGFR ≥25 to ≤75 mL/min/1.73 m2 and albuminuria (UACR ≥200 and ≤5000 mg/g) to determine the effect of dapagliflozin compared with placebo, when added to background standard of care therapy, on the incidence of the composite endpoint of ≥50% sustained decline in eGFR, end-stage kidney disease (ESKD) (defined as sustained eGFR <15 mL/min/1.73 m2, chronic dialysis treatment or receiving a renal transplant), cardiovascular or renal death.
Of 4,304 patients, 2,152 were randomised to dapagliflozin 10 mg and 2,152 to placebo and followed for a median of 28.5 months. Treatment was continued if eGFR fell to levels below 25 mL/min/1.73 m2 during the study and could be continued in cases when dialysis was needed.
The mean age of the study population was 61.8 years, 66.9% were male. At baseline, mean eGFR was 43.1 mL/min/1.73 m2 and median UACR was 949.3 mg/g, 44.1% of patients had eGFR 30 to <45 mL/min/1.73 m2 and 14.5% had eGFR <30 mL/min/1.73 m2. 67.5% of the patients had type 2 diabetes mellitus. Patients were on standard of care (SOC) therapy; 97.0% of patients were treated with an angiotensin-converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB).
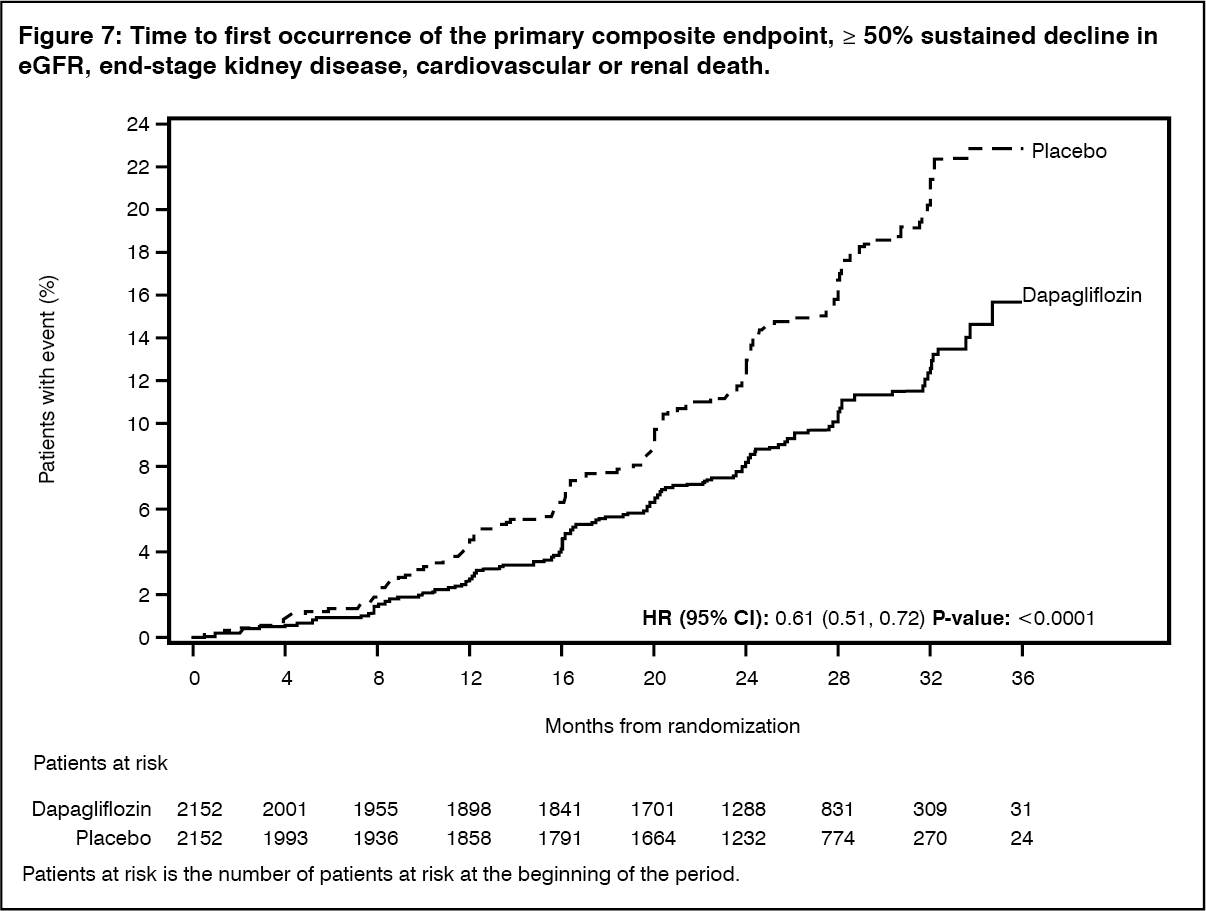
The study was stopped early for efficacy prior to the planned analysis based on a recommendation by the independent Data Monitoring Committee. Dapagliflozin was superior to placebo in preventing the primary composite endpoint of ≥50% sustained decline in eGFR, reaching end-stage kidney disease, cardiovascular or renal death. Based on the Kaplan-Meier plot for the time to first occurrence of the primary composite endpoint, the treatment effect was evident beginning at 4 months and was maintained through the end of study (Figure 7). (See Figure 7.)
 Click on icon to see table/diagram/image
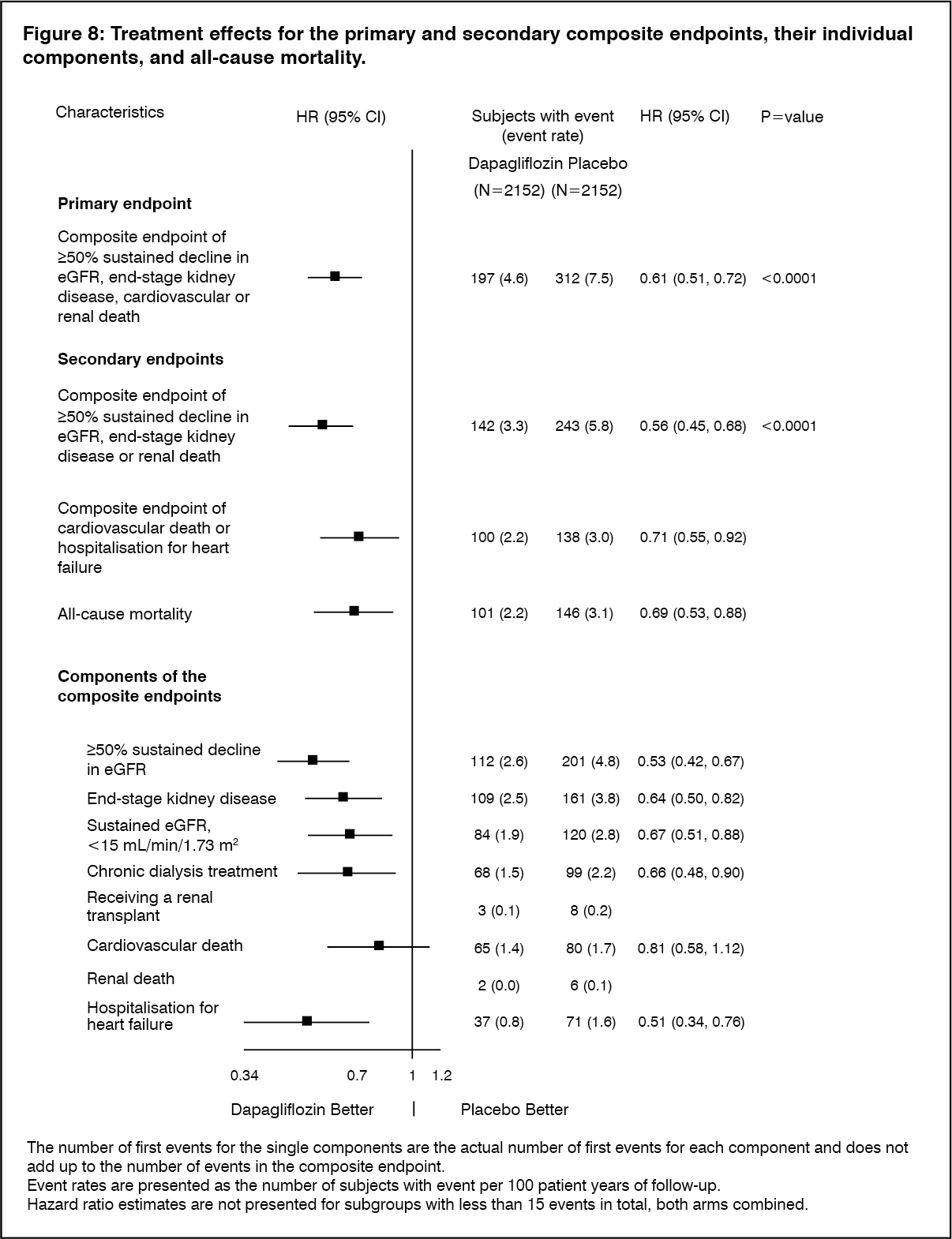
Click on icon to see table/diagram/imageAll four components of the primary composite endpoint individually contributed to the treatment effect. Dapagliflozin also reduced the incidence of the composite endpoint of ≥50% sustained decline in eGFR, end-stage kidney disease or renal death and the composite endpoint of cardiovascular death and hospitalisation for heart failure. Treatment with dapagliflozin improved overall survival in chronic kidney disease patients with a significant reduction in all-cause mortality (Figure 8). (See Figure 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe treatment benefit of dapagliflozin was consistent in chronic kidney disease patients with type 2 diabetes mellitus and without diabetes. Dapagliflozin reduced the primary composite endpoint of ≥50% sustained decline in eGFR, reaching end-stage kidney disease, cardiovascular or renal death with a HR of 0.64 (95% CI 0.52, 0.79) in patients with type 2 diabetes mellitus and 0.50 (95% CI 0.35, 0.72) in patients without diabetes.
The treatment benefit of dapagliflozin over placebo on the primary endpoint was also consistent across other key subgroups, including eGFR, age, gender, and region.
Pharmacokinetics: Absorption: Dapagliflozin was rapidly and well absorbed after oral administration. Maximum dapagliflozin plasma concentrations (Cmax) were usually attained within 2 hours after administration in the fasted state. Geometric mean steady-state dapagliflozin Cmax and AUCτ values following once daily 10 mg doses of dapagliflozin were 158 ng/mL and 628 ng h/mL, respectively. The absolute oral bioavailability of dapagliflozin following the administration of a 10 mg dose is 78%. Administration with a high-fat meal decreased dapagliflozin Cmax by up to 50% and prolonged Tmax by approximately 1 hour, but did not alter AUC as compared with the fasted state. These changes are not considered to be clinically meaningful. Hence, Forxiga can be administered with or without food.
Distribution: Dapagliflozin is approximately 91% protein bound. Protein binding was not altered in various disease states (e.g. renal or hepatic impairment). The mean steady-state volume of distribution of dapagliflozin was 118 litres.
Biotransformation: Dapagliflozin is extensively metabolised, primarily to yield dapagliflozin 3-O-glucuronide, which is an inactive metabolite. Dapagliflozin 3-O-glucuronide or other metabolites do not contribute to the glucose-lowering effects. The formation of dapagliflozin 3-O-glucuronide is mediated by UGT1A9, an enzyme present in the liver and kidney, and CYP-mediated metabolism was a minor clearance pathway in humans.
Elimination: The mean plasma terminal half-life (t½) for dapagliflozin was 12.9 hours following a single oral dose of dapagliflozin 10 mg to healthy subjects. The mean total systemic clearance of dapagliflozin administered intravenously was 207 mL/min. Dapagliflozin and related metabolites are primarily eliminated via urinary excretion with less than 2% as unchanged dapagliflozin. After administration of a 50 mg [14C]-dapagliflozin dose, 96% was recovered, 75% in urine and 21% in faeces. In faeces, approximately 15% of the dose was excreted as parent drug.
Linearity: Dapagliflozin exposure increased proportional to the increment in dapagliflozin dose over the range of 0.1 to 500 mg and its pharmacokinetics did not change with time upon repeated daily dosing for up to 24 weeks.
Special populations: Renal impairment: At steady-state (20 mg once-daily dapagliflozin for 7 days), subjects with type 2 diabetes mellitus and mild, moderate or severe renal impairment (as determined by iohexol plasma clearance) had mean systemic exposures of dapagliflozin of 32%, 60% and 87% higher, respectively, than those of subjects with type 2 diabetes mellitus and normal renal function. The steady-state 24-hour urinary glucose excretion was highly dependent on renal function and 85, 52, 18 and 11 g of glucose/day was excreted by subjects with type 2 diabetes mellitus and normal renal function or mild, moderate or severe renal impairment, respectively. The impact of hemodialysis on dapagliflozin exposure is not known. The effect of reduced renal function on systemic exposure was evaluated in a population pharmacokinetic model. Consistent with previous results, model predicted AUC was higher in patients with chronic kidney disease compared with patients with normal renal function, and was not meaningfully different in chronic kidney disease patients with type 2 diabetes mellitus and without diabetes.
Hepatic impairment: In subjects with mild or moderate hepatic impairment (Child-Pugh classes A and B), mean Cmax and AUC of dapagliflozin were up to 12% and 36% higher, respectively, compared to healthy matched control subjects. These differences were not considered to be clinically meaningful. In subjects with severe hepatic impairment (Child-Pugh class C) mean Cmax and AUC of dapagliflozin were 40% and 67% higher than matched healthy controls, respectively.
Elderly (≥65 years): There is no clinically meaningful increase in exposure based on age alone in subjects up to 70 years old. However, an increased exposure due to age-related decrease in renal function can be expected. There are insufficient data to draw conclusions regarding exposure in patients >70 years old.
Paediatric population: Pharmacokinetics in the paediatric population have not been studied.
Gender: The mean dapagliflozin AUCss in females was estimated to be about 22% higher than in males.
Race: There were no clinically relevant differences in systemic exposures between White, Black or Asian races.
Body weight: Dapagliflozin exposure was found to decrease with increased weight. Consequently, low-weight patients may have somewhat increased exposure and patients with high weight somewhat decreased exposure. However, the differences in exposure were not considered clinically meaningful.
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential and fertility. Dapagliflozin did not induce tumours in either mice or rats at any of the doses evaluated in two-year carcinogenicity studies.
Reproductive and developmental toxicity: Direct administration of dapagliflozin to weanling juvenile rats and indirect exposure during late pregnancy (time periods corresponding to the second and third trimesters of pregnancy with respect to human renal maturation) and lactation are each associated with increased incidence and/or severity of renal pelvic and tubular dilatations in progeny.
In a juvenile toxicity study, when dapagliflozin was dosed directly to young rats from postnatal day 21 until postnatal day 90, renal pelvic and tubular dilatations were reported at all dose levels; pup exposures at the lowest dose tested were ≥ 15 times the maximum recommended human dose. These findings were associated with dose-related increases in kidney weight and macroscopic kidney enlargement observed at all doses. The renal pelvic and tubular dilatations observed in juvenile animals did not fully reverse within the approximate 1-month recovery period.
In a separate study of pre- and postnatal development, maternal rats were dosed from gestation day 6 through postnatal day 21, and pups were indirectly exposed in utero and throughout lactation (a satellite study was conducted to assess dapagliflozin exposures in milk and pups). Increased incidence or severity of renal pelvic dilatation was observed in adult offspring of treated dams, although only at the highest dose tested (associated maternal and pup dapagliflozin exposures were 1,415 times and 137 times, respectively, the human values at the maximum recommended human dose). Additional developmental toxicity was limited to dose-related reductions in pup body weights, and observed only at doses ≥15 mg/kg/day (associated with pup exposures that are ≥29 times the human values at the maximum recommended human dose). Maternal toxicity was evident only at the highest dose tested, and limited to transient reductions in body weight and food consumption at dose. The no observed adverse effect level (NOAEL) for developmental toxicity, the lowest dose tested, is associated with a maternal systemic exposure multiple that is approximately 19 times the human value at the maximum recommended human dose.
In additional studies of embryo-foetal development in rats and rabbits, dapagliflozin was administered for intervals coinciding with the major periods of organogenesis in each species. Neither maternal nor developmental toxicities were observed in rabbits at any dose tested; the highest dose tested is associated with a systemic exposure multiple of approximately 1,191 times the maximum recommended human dose. In rats, dapagliflozin was neither embryo lethal nor teratogenic at exposures up to 1,441 times the maximum recommended human dose.