Monitor carefully patients for signs and symptoms of hypersensitivity reactions during and following each administration of Ferinject.
Ferinject should only be administered when staff trained to evaluate and manage anaphylactic reactions is immediately available, in an environment where full resuscitation facilities can be assured. The patient should be observed for adverse effects for at least 30 minutes following each Ferinject administration (see Precautions).
Posology: The posology of Ferinject follows a stepwise approach: [1] determination of the individual iron need, [2] calculation and administration of the iron dose(s), and [3] post-iron repletion assessments.These steps are outlined as follows:
Step 1: Determination of the iron need: The individual iron need for repletion using Ferinject is determined based on the patient's body weight and haemoglobin (Hb) level. Refer to Table 1 for determination of the iron need: (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Iron deficiency must be confirmed by laboratory tests as stated in Indications/Uses.
Step 2: Calculation and administration of the maximum individual iron dose(s): Based on the iron need determined previously, the appropriate dose(s) of Ferinject should be administered taking into consideration the following: A single Ferinject administration should not exceed: 15 mg iron/kg body weight (for administration by intravenous injection) or 20 mg iron/kg body weight (for administration by intravenous infusion); 1,000 mg of iron (20 mL Ferinject).
The maximum recommended cumulative dose of Ferinject is 1,000 mg of iron (20 mL Ferinject) per week.
Step 3: Post-iron repletion assessments: Re-assessment should be performed by the clinician based on the individual patient's condition. The Hb level should be re-assessed no earlier than 4 weeks post final Ferinject administration to allow adequate time for erythropoiesis and iron utilisation. In the event the patient requires further iron repletion, the iron need should be recalculated using Table 1 previously. (See Pharmacology: Pharmacodynamics under Actions.)
Special Population - patients with haemodialysis-dependent chronic kidney disease: A single maximum daily dose of 200 mg iron should not be exceeded in haemodialysis-dependent chronic kidney disease patients (see also Precautions).
Paediatric population: The use of Ferinject has not been studied in children, and therefore is not recommended in children under 14 years.
Method of administration: Ferinject must only be administered by the intravenous route: by injection, or by infusion, or during a haemodialysis session undiluted directly into the venous limb of the dialyser.
Ferinject must not be administered by the subcutaneous or intramuscular route.
Intravenous injection: Ferinject may be administered by intravenous injection using undiluted solution. The maximum single dose is 15 mg iron/kg body weight but should not exceed 1,000 mg iron. The administration rates are as shown in Table 2: (See Table 2.)
Click on icon to see table/diagram/image
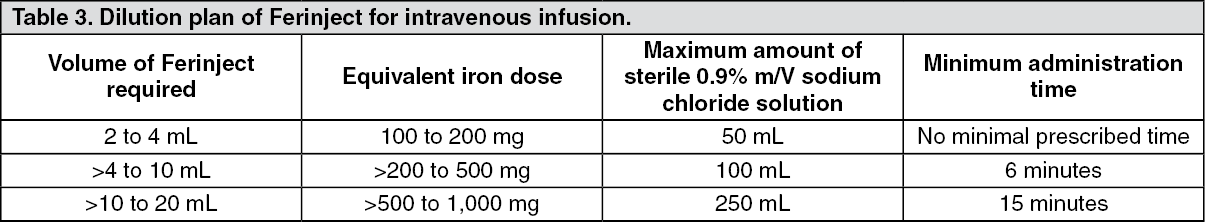
Intravenous infusion: Ferinject may be administered by intravenous infusion, in which case it must be diluted. The maximum single dose is 20 mg iron/kg body weight, but should not exceed 1,000 mg iron.
For infusion, Ferinject must only be diluted in sterile 0.9% m/V sodium chloride solution as shown in Table 3.
Note: for stability reasons, Ferinject should not be diluted to concentrations less than 2 mg iron/mL (not including the volume of the ferric carboxymaltose solution). For further instructions on dilution of the medicinal product before administration, see Special precautions for disposal and other handling under Cautions for Usage. (See Table 3.)
Click on icon to see table/diagram/image


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
