Renal function: There is data on use of deferasirox only in patients with baseline serum creatinine within the age-appropriate normal range.
Increases in serum creatinine of >33% on ≥2 consecutive occasions, sometimes above the upper limit of the normal range, occurred. These were dose-dependent. About two-thirds of the patients showing serum creatinine increase returned below the 33% level without dose adjustment. In the remaining third the serum creatinine increase did not always respond to a dose reduction or a dose interruption. In some cases, only a stabilisation of the serum creatinine values has been observed after dose reduction. Cases of acute renal failure have been reported following post-marketing use of deferasirox (see Adverse Reactions). In some post-marketing cases, renal function deterioration has led to renal failure requiring temporary or permanent dialysis.
The causes of the rises in serum creatinine have not been elucidated. Particular attention should therefore be paid to monitoring of serum creatinine in patients who are concomitantly receiving medicinal products that depress renal function, and in patients who are receiving high doses of deferasirox and/or low rates of transfusion (<7 ml/kg/month of packed red blood cells or <2 units/month for an adult). While no increase in renal adverse events was observed after dose escalation of Deferasirox dispersible tablets to doses above 30 mg/kg, an increased risk of renal adverse events with film-coated tablet doses above 21 mg/kg cannot be excluded.
It is recommended that serum creatinine be assessed in duplicate before initiating therapy. Serum creatinine, creatinine clearance (estimated with the Cockcroft-Gault or MDRD formula in adults and with the Schwartz formula in children) and/or plasma cystatin C levels should be monitored prior to therapy, weekly in the first month after initiation or modification of therapy with FERASIRO (including switch of formulation), and monthly thereafter. Patients with pre-existing renal conditions and patients who are receiving medicinal products that depress renal function may be more at risk of complications. Care should be taken to maintain adequate hydration in patients who develop diarrhoea or vomiting.
There have been post-marketing reports of metabolic acidosis occurring during treatment with deferasirox. The majority of these patients had renal impairment, renal tubulopathy (Fanconi syndrome) or diarrhoea, or conditions where acid-base imbalance is a known complication. Acid-base balance should be monitored as clinically indicated in these populations. Interruption of FERASIRO therapy should be considered in patients who develop metabolic acidosis.
Post-marketing cases of severe forms of renal tubulopathy (such as Fanconi syndrome) and renal failure associated with changes in consciousness in the context of hyperammonaemic encephalopathy have been reported in patients treated with deferasirox, mainly in children. It is recommended that hyperammonaemic encephalopathy be considered and ammonia levels measured in patients who develop unexplained changes in mental status while on Ferasiro therapy. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Treatment may be reinitiated depending on the individual clinical circumstances.
Dose reduction or interruption may be also considered if abnormalities occur in levels of markers of renal tubular function and/or as clinically indicated: Proteinuria (test should be performed prior to therapy and monthly thereafter); Glycosuria in non-diabetics and low levels of serum potassium, phosphate, magnesium or urate, phosphaturia, aminoaciduria (monitor as needed).
Renal tubulopathy has been mainly reported in children and adolescents with beta-thalassaemia treated with FERASIRO.
Patients should be referred to a renal specialist, and further specialised investigations (such as renal biopsy) may be considered if the following occur despite dose reduction and interruption: Serum creatinine remains significantly elevated and Persistent abnormality in another marker of renal function (e.g. proteinuria, Fanconi Syndrome).
Hepatic function: Liver function test elevations have been observed in patients treated with deferasirox. Post-marketing cases of hepatic failure, some of which were fatal, have been reported. Severe forms associated with changes in consciousness in the context of hyperammonaemic encephalopathy, may occur in patients treated with deferasirox, particularly in children. It is recommended that hyperammonaemic encephalopathy be considered and ammonia levels measured in patients who develop unexplained changes in mental status while on Ferasiro therapy. Care should be taken to maintain adequate hydration in patients who experience volume-depleting events (such as diarrhoea or vomiting), particularly in children with acute illness. Most reports of hepatic failure involved patients with significant comorbidities including pre-existing chronic liver conditions (including cirrhosis and hepatitis C) and multi-organ failure. The role of deferasirox as a contributing or aggravating factor cannot be excluded (see Adverse Reactions).
It is recommended that serum transaminases, bilirubin and alkaline phosphatase be checked before the initiation of treatment, every 2 weeks during the first month and monthly thereafter. If there is a persistent and progressive increase in serum transaminase levels that cannot be attributed to other causes, FERASIRO should be interrupted. Once the cause of the liver function test abnormalities has been clarified or after return to normal levels, cautious re-initiation of treatment at a lower dose followed by gradual dose escalation may be considered.
FERASIRO is not recommended in patients with severe hepatic impairment (Child-Pugh Class C) (see Pharmacology: Pharmacokinetics under Actions). (See Table 5.)
Click on icon to see table/diagram/image
In patients with a short life expectancy (e.g. high-risk myelodysplastic syndromes), especially when co-morbidities could increase the risk of adverse events, the benefit of FERASIRO might be limited and may be inferior to risks. As a consequence, treatment with FERASIRO is not recommended in these patients.
Caution should be used in elderly patients due to a higher frequency of adverse reactions (in particular, diarrhoea).
Data in children with non-transfusion-dependent thalassaemia are very limited (see Pharmacology: Pharmacodynamics under Actions). As a consequence, FERASIRO therapy should be closely monitored to detect adverse reactions and to follow iron burden in the paediatric population. In addition, before treating heavily iron-overloaded children with non-transfusion-dependent thalassaemia with FERASIRO, the physician should be aware that the consequences of long-term exposure in such patients are currently not known.
Gastrointestinal disorders: Upper gastrointestinal ulceration and haemorrhage have been reported in patients, including children and adolescents, receiving deferasirox. Multiple ulcers have been observed in some patients (see Adverse Reactions). There have been reports of ulcers complicated with digestive perforation. Also, there have been reports of fatal gastrointestinal haemorrhages, especially in elderly patients who had haematological malignancies and/or low platelet counts. Physicians and patients should remain alert for signs and symptoms of gastrointestinal ulceration and haemorrhage during FERASIRO therapy. In case of gastrointestinal ulceration or haemorrhage, FERASIRO should be discontinued and additional evaluation and treatment must be promptly initiated. Caution should be exercised in patients who are taking FERASIRO in combination with substances that have known ulcerogenic potential, such as NSAIDs, corticosteroids, or oral bisphosphonates, in patients receiving anticoagulants and in patients with platelet counts below 50,000/mm
3 (50 x 10
9/l) (see Interactions).
Skin disorders: Skin rashes may appear during FERASIRO treatment. The rashes resolve spontaneously in most cases. When interruption of treatment may be necessary, treatment may be reintroduced after resolution of the rash, at a lower dose followed by gradual dose escalation. In severe cases this reintroduction could be conducted in combination with a short period of oral steroid administration. Severe cutaneous adverse reactions (SCARs) including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) and drug reaction with eosinophilia and systemic symptoms (DRESS), which could be life-threatening or fatal, have been reported. If any SCAR is suspected, FERASIRO should be discontinued immediately and should not be reintroduced. At the time of prescription, patients should be advised of the signs and symptoms of severe skin reactions, and be closely monitored.
Hypersensitivity reactions: Cases of serious hypersensitivity reactions (such as anaphylaxis and angioedema) have been reported in patients receiving deferasirox, with the onset of the reaction occurring in the majority of cases within the first month of treatment (see Adverse Reactions). If such reactions occur, FERASIRO should be discontinued and appropriate medical intervention instituted. Deferasirox should not be reintroduced in patients who have experienced a hypersensitivity reaction due to the risk of anaphylactic shock (see Contraindications).
Vision and hearing: Auditory (decreased hearing) and ocular (lens opacities) disturbances have been reported (see Adverse Reactions). Auditory and ophthalmic testing (including fundoscopy) is recommended before the start of treatment and at regular intervals thereafter (every 12 months). If disturbances are noted during the treatment, dose reduction or interruption may be considered.
Blood disorders: There have been post-marketing reports of leukopenia, thrombocytopenia or pancytopenia (or aggravation of these cytopenias) and of aggravated anaemia in patients treated with deferasirox. Most of these patients had pre-existing haematological disorders that are frequently associated with bone marrow failure. However, a contributory or aggravating role cannot be excluded. Interruption of treatment should be considered in patients who develop unexplained cytopenia.
Other considerations: Monthly monitoring of serum ferritin is recommended in order to assess the patient's response to therapy and to avoid overchelation (see Dosage & Administration). Dose reduction or closer monitoring of renal and hepatic function, and serum ferritin levels are recommended during periods of treatments with high doses and when serum ferritin levels are close to the target range. If serum ferritin falls consistently below 500 μg/l (in transfusional iron overload) or below 300 μg/l (in non-transfusion-dependent thalassaemia syndromes), an interruption of treatment should be considered.
The results of the tests for serum creatinine, serum ferritin and serum transaminases should be recorded and regularly assessed for trends.
Growth and sexual development of paediatric patients treated with deferasirox for up to 5 years were not affected (see Adverse Reactions). However, as a general precautionary measure in the management of paediatric patients with transfusional iron overload, body weight, height and sexual development should be monitored prior to therapy and at regular intervals (every 12 months).
Cardiac dysfunction is a known complication of severe iron overload. Cardiac function should be monitored in patients with severe iron overload during long-term treatment with FERASIRO.
Effects on ability to drive and use machines: FERASIRO has minor influence on the ability to drive and use machines. Patients experiencing the uncommon adverse reaction of dizziness should exercise caution when driving or operating machines (see Adverse Reactions).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image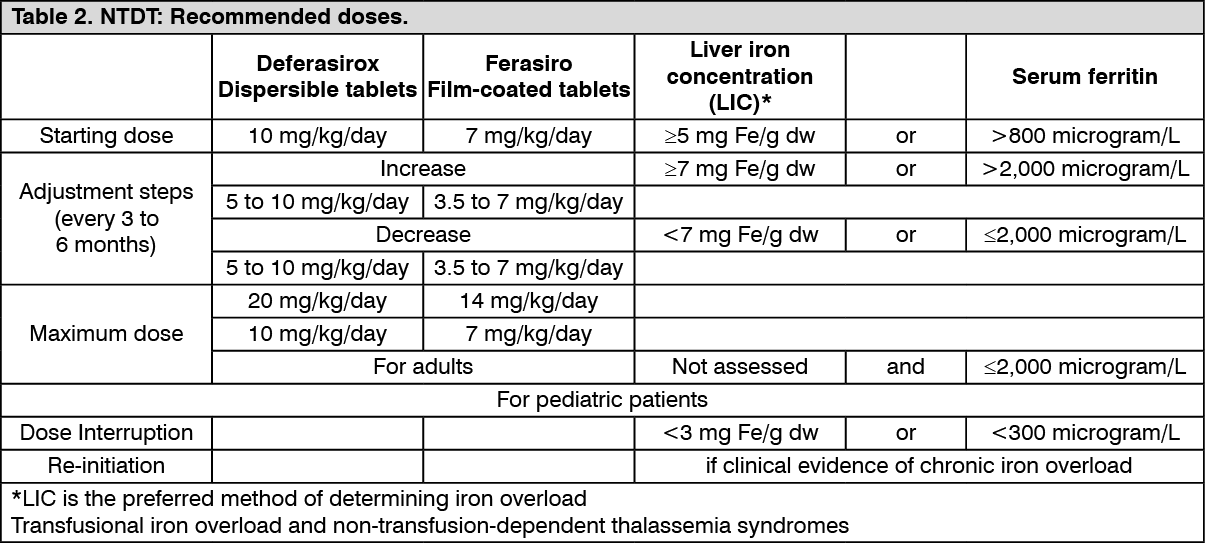 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
