Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trial of another drug and may not reflect the rates observed in patients in clinical practice.
The most serious adverse reactions reported with Fabrazyme treatment during clinical trials were anaphylactic and allergic reactions (see Infusion Reactions under Warnings).
The most common adverse reactions reported with Fabrazyme are infusion reactions, some of which were severe (see Infusion Reactions under Warnings). Serious and/or frequently occurring (≥5% incidence) related adverse reactions, consisted of one or more of the following: chills, pyrexia, feeling hot or cold, dyspnea, nausea, flushing, headache, vomiting, paresthesia, fatigue, pruritus, pain in extremity, hypertension, chest pain, throat tightness, abdominal pain, dizziness, tachycardia, nasal congestion, diarrhea, edema peripheral, myalgia, back pain, pallor, bradycardia, urticaria, hypotension, face edema, rash, and somnolence. The occurrence of somnolence can be attributed to clinical trial specified pre-treatment with antihistamines. Most infusion-related reactions requiring intervention were ameliorated with slowing of the infusion rate, temporarily stopping the infusion, and/or administration of antipyretics, antihistamines, or steroids.
Other reported serious adverse events included stroke, pain, ataxia, bradycardia, cardiac arrhythmia, cardiac arrest, decreased cardiac output, vertigo, hypoacousia and nephrotic syndrome. These adverse events also occur as manifestations of Fabry disease; an alteration in frequency or severity cannot be determined from the small numbers of patients studied.
The data described as follows reflect exposure of 80 patients, ages 16 to 61 years, to 1.0 mg/kg Fabrazyme every two weeks in two separate double-blind, placebo-controlled clinical trials, for periods ranging from 1 to 35 months (mean 15.5 months). All 58 patients enrolled in one of the two studies continued into an open-label extension study of Fabrazyme treatment for up to 54 additional months. Patients were treated with antipyretics and antihistamines prior to the infusions.
Table 3 enumerates treatment-emergent adverse events (regardless of relationship) that occurred during the double-blind treatment periods of the two placebo-controlled trials (Study 1 and Study 2) (See Pharmacology: Pharmacodynamics: Clinical Studies under Actions). Reported adverse events have been classified by Medical Dictionary for Regulatory Activities (MedDRA) terminology System Organ Class and Preferred Term. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Observed adverse events in the Phase 1/2 study and the open-label treatment period for the extension study following the controlled study were not different in nature or intensity.
The safety profile of Fabrazyme in pediatric Fabry disease patients, ages 8 to 16 years, was found to be consistent with that seen in adults (see Pharmacology: Pharmacodynamics: Clinical Studies under Actions and Pediatric Use under Precautions). The safety of Fabrazyme in patients younger than 8 years of age has not been evaluated.
Immunogenicity: Ninety-five of 121 (79%) adult patients and 11 of 16 (69%) pediatric patients (106 of 137, 74% of all patients) treated with Fabrazyme in clinical studies have developed IgG antibodies to Fabrazyme. Most patients who develop IgG antibodies do so within the first three months of exposure. IgG seroconversion in pediatric patients was associated with prolonged half-life of Fabrazyme, a phenomenon rarely observed in adult patients (see Pharmacokinetics under Actions and Pediatric Use under Precautions). A possible cause for this prolongation likely pertains to the ability of antibodies to act as "carriers" for their antigens. Among the 14 female patients exposed to Fabrazyme in clinical studies, six (adult patients) developed IgG antibodies to Fabrazyme.
IgG antibodies to Fabrazyme were purified from 15 patients with high antibody titers (≥12,800) and studied for inhibition of
in vitro enzyme activity. Under the conditions of this assay, most of these 15 patients had inhibition of
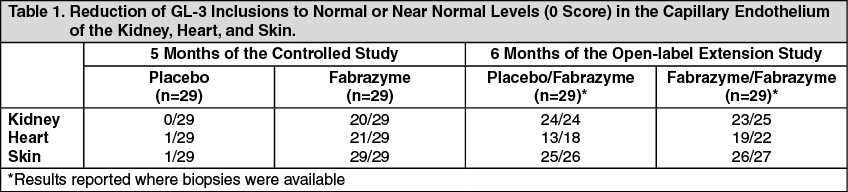
in vitro enzyme activity ranging between 21-74% at one or more time points during the study. Assessment of inhibition of enzyme uptake in cells has not been performed. No general pattern was seen in individual patient reactivity over time. The clinical significance of binding and/or inhibitory antibodies to Fabrazyme is not known. In patients followed in the open-label extension study, reduction of GL-3 in plasma and GL-3 inclusions in superficial skin capillaries was maintained after antibody formation.
As with all therapeutic proteins, there is potential for immunogenicity. The data reflect the percentage of patients whose test results were considered positive for antibodies to Fabrazyme using an ELISA and radioimmunoprecipitation (RIP) assay for antibodies.
The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibodies (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of the incidence of antibodies to Fabrazyme with the incidence of antibodies to other products may be misleading.
Testing for IgE antibodies was performed in approximately 60 patients in clinical trials who experienced moderate to severe infusion reactions or in whom mast cell activation was suspected. Seven of these patients tested positive for Fabrazyme-specific IgE antibodies or had a positive skin test to Fabrazyme. Patients who have had a positive skin test to Fabrazyme, or who have tested positive for Fabrazyme-specific IgE antibodies in clinical trials with Fabrazyme have been rechallenged (see Pharmacology: Pharmacodynamics: Clinical Studies under Actions, Dosage & Administration and Immunogenicity and Rechallenge under Precautions).
Postmarketing Experience: The following adverse reactions have been identified during post approval use of FABRAZYME. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In postmarketing experience with agalsidase beta, severe and serious infusion-related reactions have been reported, some of which were life-threatening, including anaphylactic shock (see Infusion Reactions under Warnings and Precautions). Reactions have included localized angioedema (including auricular swelling, eye swelling, dysphagia, lip swelling, edema, pharyngeal edema, face swelling, and swollen tongue), generalized urticaria, bronchospasm, and hypotension.
Adverse reactions (regardless of relationship) resulting in death reported in the postmarketing setting with FABRAZYME treatment included cardiorespiratory arrest, respiratory failure, cardiac failure, sepsis, cerebrovascular accident, myocardial infarction, renal failure, and pneumonia. Some of these reactions were reported in Fabry disease patients with significant underlying disease.
In addition to the adverse reactions reported in ADVERSE REACTIONS the following adverse reactions have been reported during postmarketing use of agalsidase beta: arthralgia, asthenia, erythema, hyperhidrosis, infusion site reaction, lacrimation increased, leukocytoclastic vasculitis, lymphadenopathy, hypoesthesia, oral hypoesthesia, palpitations, rhinorrhea, oxygen saturation decreased, and hypoxia.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image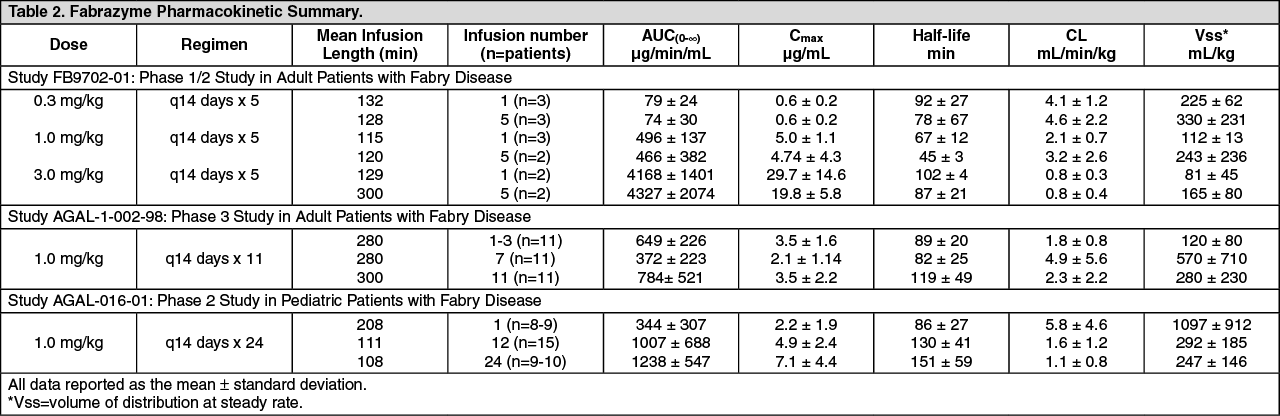 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image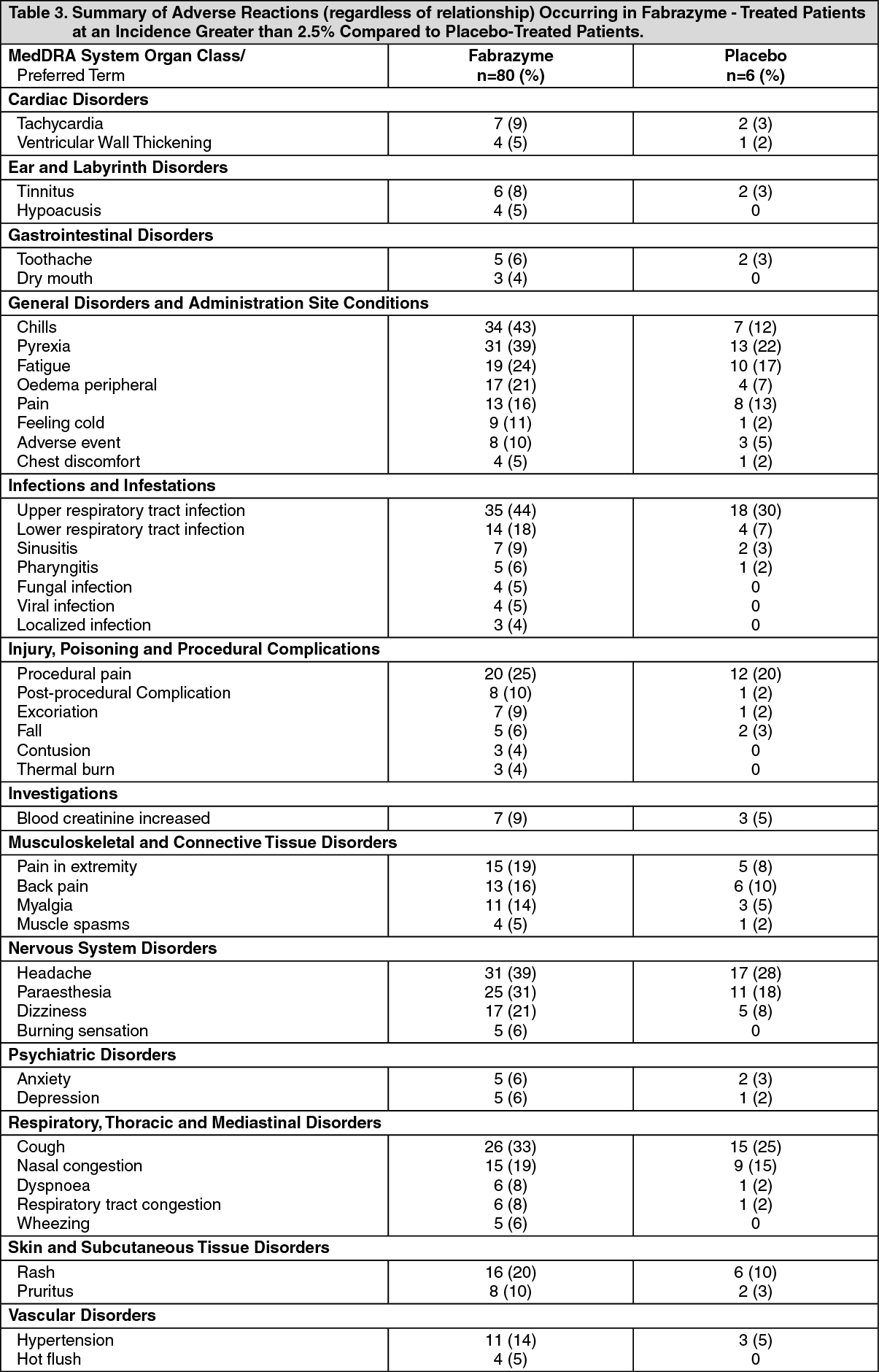 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
