Pharmacotherapeutic group: Other drugs affecting bone structure and mineralisation.
ATC code: M05BX06.
Pharmacology: Pharmacodynamics: Mechanism of Action: Romosozumab is a humanised monoclonal antibody (IgG2) that binds and inhibits sclerostin. Romosozumab has a dual effect on bone, increasing bone formation and decreasing bone resorption. Romosozumab stimulates new bone formation on trabecular and endocortical bone surfaces by stimulating osteoblastic activity resulting in increases in trabecular and cortical bone mass and improvements in bone structure and strength.
Pharmacodynamic Effects: EVENITY has a dual effect on bone, increasing bone formation and decreasing bone resorption. In postmenopausal women with osteoporosis, EVENITY increased the bone formation marker procollagen type 1 N-terminal propeptide (P1NP) early in treatment, with a peak increase of approximately 145% relative to placebo 2 weeks after initiating treatment, followed by a return to placebo levels at month 9 and a decline to approximately 15% below placebo at month 12. EVENITY decreased the bone resorption marker type 1 collagen C-telopeptide (CTX) with a maximal reduction of approximately 55% relative to placebo 2 weeks after initiating treatment. CTX levels remained below placebo and were approximately 25% below placebo at month 12.
After discontinuation of EVENITY therapy in postmenopausal women with osteoporosis, P1NP levels returned to baseline within 12 months; CTX increased above baseline levels within 3 months and returned toward baseline levels by month 12, reflecting reversibility of effect. Upon retreatment with EVENITY after 12 months off treatment, the level of increase in P1NP and decrease in CTX by EVENITY was similar to that observed during the initial treatment.
In women transitioning from oral alendronate, EVENITY also increased bone formation and decreased bone resorption.
Clinical Data: Treatment of Osteoporosis in Postmenopausal Women: Study 1 (ARCH, alendronate-controlled) was a randomised, double blind, alendronate-controlled study of 4093 postmenopausal women aged 55 to 90 years (mean age of 74.3 years), with a median follow-up of 33 months.
The mean baseline lumbar spine, total hip, and femoral neck BMD T-scores were -2.96, -2.80, and -2.90, respectively, 96.1% of women had a vertebral fracture at baseline, and 99.8% of women had a previous fracture. Women were randomised (1:1) to receive either monthly subcutaneous injections of EVENITY (N = 2046) or oral weekly alendronate (N = 2047) for 12 months, with daily supplementation of calcium and vitamin D. After the 12-month treatment period, women in both arms transitioned to open-label alendronate while remaining blinded to their initial treatment. The primary analysis was performed when all women had completed the month 24 study visit and clinical fracture events were confirmed for at least 330 women, which occurred after a median of 33 months on study.
The primary efficacy endpoints were the incidence of new vertebral fracture through month 24 and the incidence of clinical fracture (defined as the composite of nonvertebral fracture and clinical vertebral fracture) at primary analysis. Secondary efficacy endpoints included the incidence of nonvertebral fractures, hip fractures, and major nonvertebral fractures at the primary analysis, and percent change from baseline in BMD at the lumbar spine, total hip, and femoral neck at month 12 and month 24.
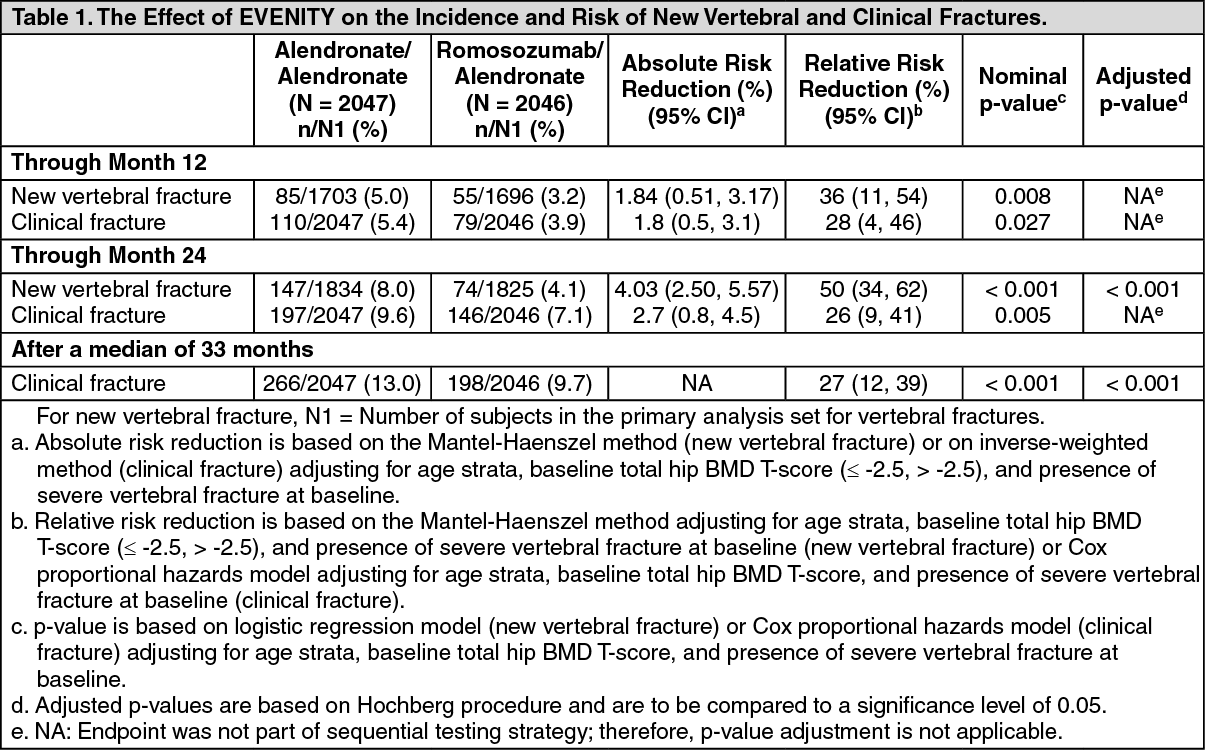
Effect on New Vertebral and Clinical Fractures: EVENITY reduced the incidence of new vertebral fracture at 24 months and clinical fracture after a median of 33 months. The number of patients who experienced vertebral and clinical fracture was consistently lower in the EVENITY arm at prespecified time points. See Table 1 for full data. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
EVENITY for 12 months followed by alendronate for 12 months demonstrated a persistent effect in reducing the incidence of new vertebral fractures (see Figure 1).
Click on icon to see table/diagram/image
Effect on Other Fracture Types/Groups: EVENITY significantly reduced the incidence of nonvertebral fracture after a median follow up of 33 months. EVENITY reduced the number of patients who experienced nonvertebral fracture, hip fracture, and major nonvertebral fractures compared to alendronate consistently at prespecified time points. See Table 2 for full data. (See Table 2.)
Click on icon to see table/diagram/image
The Kaplan-Meier estimates of the cumulative incidence of clinical fracture, nonvertebral fracture and hip fracture over time are shown in Figure 2 as follows. (See Figure 2).
Click on icon to see table/diagram/image
Effect on Bone Mineral Density (BMD): EVENITY significantly increased BMD at the lumbar spine, total hip, and femoral neck compared with alendronate at month 12. At month 24, 12-month treatment with EVENITY followed by 12-month treatment with alendronate significantly increased BMD compared with alendronate alone for 24 months at the lumbar spine, total hip, and femoral neck. The BMD increase with EVENITY over alendronate observed at month 12 was maintained at month 24 (see Table 3).
Click on icon to see table/diagram/image
A total of 167 subjects participated in the imaging component (as measured by DXA) of the Imaging and PK/BTM/Biomarker Substudy. EVENITY resulted in progressive increases from baseline in BMD beginning at month 6. Treatment differences in BMD between EVENITY and alendronate groups continued to increase at month 12. After transitioning to alendronate after 12-month treatment with EVENITY, treatment differences in BMD between the EVENITY-alendronate and alendronate-alendronate groups continued to increase at month 18 and were maintained at month 24 (see Figure 3).
Consistent effects on BMD were observed regardless of baseline age, baseline BMD, and geographic region at the lumbar spine, total hip, and femoral neck.
Treatment differences in BMD at 6 months were 7.6% at the lumbar spine, 2.2% at the total hip, and 2.9% at the femoral neck. At 12 months, the treatment differences were 8.9% at the lumbar spine, 3.7% at the total hip, and 4.1% at the femoral neck. At 18 months, after transitioning to alendronate after 12 months of EVENITY treatment, the treatment differences between the EVENITY-alendronate and alendronate-alendronate groups were 9.3% at the lumbar spine, 4.3% at the total hip, and 5.4% at the femoral neck. At 24 months, the EVENITY-alendronate group maintained gains in BMD compared to on the alendronate-alendronate group, with treatment differences of 9.4% at the lumbar spine, 4.3% at the total hip, and 5.3% at the femoral neck. (See Figure 3.)
Click on icon to see table/diagram/image
Study 2 (FRAME, placebo-controlled) was a randomised, double blind, placebo-controlled study of 7180 postmenopausal women aged 55 to 90 years (mean age of 70.9 years). The mean baseline lumbar spine, total hip, and femoral neck BMD T-scores were -2.72, -2.47, and -2.75, respectively, and 18.3% of women had a vertebral fracture at baseline.
Women were randomised to receive subcutaneous injections of either EVENITY (N = 3589) or placebo (N = 3591) once every month for 12 months with daily supplementation of calcium and vitamin D. After the 12-month treatment period, women in both arms transitioned to open-label denosumab 60 mg subcutaneous every 6 months for 12 months while remaining blinded to initial treatment.
The co-primary efficacy endpoints were the incidence of new vertebral fractures through month 12 and through month 24. Secondary efficacy endpoints included the incidence of clinical fractures (all symptomatic fractures including nonvertebral and painful vertebral fractures), nonvertebral fractures, new or worsening vertebral fractures, major nonvertebral fractures, hip fractures, and percent change from baseline in BMD at the lumbar spine, total hip, and femoral neck, and were evaluated though 24 months.
Effect on New Vertebral and Clinical Fractures: EVENITY significantly reduced the incidence of new vertebral fractures through month 12 (p < 0.001), as shown in Table 4. The reduction in fracture risk persisted through the second year in women who received EVENITY during the first year and transitioned to denosumab compared to those who transitioned from placebo to denosumab (month 24; p < 0.001).
EVENITY also significantly reduced the incidence of clinical fractures through month 12 (see Table 4 and Figure 4 for time to first clinical fracture). (See Table 4 and Figures 4 and 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Effect on Other Fracture Types/Groups: See Table 5 for effect of EVENITY on other Fracture Types/Groups through month 24. (See Table 5).
Click on icon to see table/diagram/image
Effect on Bone Mineral Density (BMD): EVENITY significantly increased BMD at the lumbar spine, total hip, and femoral neck compared to placebo at month 12. Following 12 months of treatment, EVENITY increased BMD at the lumbar spine from baseline in 99% of postmenopausal women. Ninety-two percent of women treated with EVENITY achieved at least a 5% increase from baseline in BMD at lumbar spine by month 12 and 68% gained 10% or more.
These effects were sustained with transition to another osteoporosis treatment; women who received EVENITY for 12 months followed by denosumab for 12 months had greater increases in BMD at the lumbar spine, total hip, and femoral neck at month 24 compared to women who received placebo for 12 months followed by denosumab for 12 months (see Table 6).
Consistent effects on BMD were observed regardless of baseline age, baseline BMD, and geographic region at the lumbar spine, total hip, and femoral neck. (See Table 6).
Click on icon to see table/diagram/image
Among women with BMD assessed at baseline and every 6 months, EVENITY significantly increased BMD at the lumbar spine, total hip, and femoral neck relative to placebo at 6 and 12 months. Following the transition from EVENITY to denosumab, BMD continued to increase through month 24. In women who transitioned from placebo to denosumab, BMD also increased with denosumab use. The differences in BMD achieved at month 12 between EVENITY and placebo patients were overall maintained at month 24, when comparing patients who transitioned from EVENITY to denosumab versus patients who transitioned from placebo to denosumab (see Figure 6).
Treatment differences in BMD at 6 months were 9.4% at the lumbar spine, 4.3% at the total hip, and 3.6% at the femoral neck. After 12 months, the treatment differences were 13.3% at the lumbar spine, 6.9% at the total hip, and 5.9% at the femoral neck. At 18 months, women who received EVENITY followed by denosumab maintained gains in BMD compared to women who received placebo followed by denosumab, with treatment differences of 11.8% at the lumbar spine, 6.8% at the total hip, and 6.8% at the femoral neck. At 24 months, women who received EVENITY followed by denosumab maintained gains in BMD compared to women who received placebo followed by denosumab, with treatment differences of 12.6% at the lumbar spine, 6.0% at the total hip, and 6.0% at the femoral neck. (See Figure 6.)
Click on icon to see table/diagram/image
Bone Histology and Histomorphometry: A total of 154 transiliac crest bone biopsy specimens were obtained from 139 postmenopausal women with osteoporosis at month 2, month 12, and/or month 24. Of the biopsies obtained, 154 (100.0%) were adequate for qualitative histology and 138 (89.6%) were adequate for full quantitative histomorphometry assessment. Qualitative histology assessments from those treated with EVENITY showed normal bone architecture and quality at all time points. There was no evidence of woven bone, mineralisation defects, or marrow fibrosis.
Histomorphometry assessments on biopsies at months 2 and 12 compared the effect of EVENITY with placebo (15 specimens at month 2 and 39 specimens at month 12 in the EVENITY group, 14 specimens at month 2 and 31 specimens at month 12 in the placebo group). At month 2 in women treated with EVENITY, histomorphometric indices of bone formation at trabecular and endocortical surfaces were increased due to a significant increase in modelling based formation with no significant effect on remodelling formation. These effects on bone formation were accompanied by a decrease in indices of bone resorption. At month 12, both bone formation and resorption indices were decreased with EVENITY, while bone volume and trabecular and cortical thickness were increased. Biopsies obtained at month 24 compared the effect of EVENITY for 12 months followed by denosumab for 12 months (18 specimens) with placebo followed by denosumab (21 specimens). At month 24, indices of bone remodelling were low and similar in both groups, consistent with the effects of denosumab.
Treatment of Osteoporosis in Women Transitioning from Bisphosphonate Therapy: Study 3 (STRUCTURE) was a randomised, open-label study of 436 postmenopausal women aged 56 to 90 years (mean age of 71.5 years) with osteoporosis transitioning from bisphosphonate therapy to EVENITY. This study evaluated safety and BMD changes by dual-energy X-ray absorptiometry (DXA) through 12 months of treatment with EVENITY compared with 12 months of treatment with teriparatide. The study also evaluated hip strength estimated by finite element analysis (FEA) over 12 months using quantitative computed tomography images.
Enrolled women had a mean baseline lumbar spine, total hip, and femoral neck BMD T-scores of -2.85, -2.24, and -2.46, respectively, and a history of nonvertebral fracture after age 50 or vertebral fracture at any time.
At month 12, EVENITY increased BMD from baseline by 9.8% (95% CI: 9.0, 10.5) at the lumbar spine, 2.9% (95% CI: 2.5, 3.4) at the total hip, and 3.2% (95% CI: 2.6, 3.8) at the femoral neck. Treatment differences in BMD at 12 months compared to teriparatide were 4.4% (95% CI: 3.4, 5.4) at the lumbar spine, 3.4% (95% CI: 2.8, 4.0) at the total hip, and 3.4% at the femoral neck (95% CI: 2.6, 4.2; p-value < 0.0001 for all comparisons).
At month 12, EVENITY increased estimated strength from baseline by 2.5% (95% CI: 1.7, 3.2) at the total hip. The treatment difference in estimated strength at the total hip at month 12 compared to teriparatide was 3.2% (95% CI: 2.1, 4.3; p-value < 0.0001).
Adverse reactions observed in this study were generally consistent with those seen in women not transitioning from bisphosphonate therapy (discussed in Adverse Reactions).
Pharmacokinetics: Following SC administration, romosozumab exhibits nonlinear pharmacokinetics as a result of binding to sclerostin. Dose proportional increases in exposure were observed for the doses of 140 mg and higher.
Administration of a single dose of 210 mg romosozumab in healthy male and female subjects (N = 90, age range: 21 to 65 years) resulted in a mean (standard deviation [SD]) maximum serum concentration (C
max) of 22.2 (5.8) mcg/mL and a mean area under the concentration-time curve (AUC) of 389 (127) mcg*day/mL. The median time to maximum romosozumab concentration (T
max) was 5 days (range: 2 to 7 days).
Following a 210 mg subcutaneous dose, bioavailability was 81%. After C
max, serum levels declined with a mean effective half-life of 12.8 days. Steady state was generally reached by month 3 with minimal accumulation (less than 2-fold) following monthly dosing.
The presence of anti-romosozumab binding antibodies decreased romosozumab exposure up to 22%, which was not considered clinically meaningful [see Adverse Reactions].
Based on a population pharmacokinetic analysis, age (20-89 years), gender, race, or disease state (low bone mass or osteoporosis) had no clinically meaningful effects on pharmacokinetics (< 20% change in exposure at steady state). Romosozumab exposure decreased with increasing body weight. This decrease had a minimal impact on lumbar spine BMD gain (< 15% change) based on exposure response analyses and was not considered clinically meaningful. Thus, no dose adjustment is necessary based on age, gender, race, disease state, or body weight.
The pharmacokinetics of romosozumab were similar in patients transitioning from bisphosphonate therapy.
Drug Interactions: No drug-drug interaction studies have been conducted with romosozumab.
Special Populations: Paediatrics: The pharmacokinetics of romosozumab in paediatric patients have not been assessed.
Gender: The pharmacokinetics of romosozumab were similar in postmenopausal women and in men with osteoporosis.
Geriatrics: The pharmacokinetics of romosozumab were not affected by age from 20 to 89 years.
Renal Impairment: Following a single 210 mg dose of romosozumab in a clinical study of 16 patients with severe renal impairment (eGFR 15 to 29 mL/min/1.73 m
2) or end-stage renal disease (ESRD) requiring haemodialysis, mean C
max and AUC were 29% and 44% higher in patients with severe renal impairment as compared to healthy subjects. Mean romosozumab exposure was similar between patients with ESRD requiring haemodialysis and healthy subjects.
A population pharmacokinetic analysis indicated an increase in romosozumab exposure with increasing severity of renal impairment. However, based on both the renal impairment study and population PK analysis, this increase is not clinically meaningful and no dose adjustment is necessary in these patients [see Precautions].
Hepatic Impairment: No clinical studies have been conducted to evaluate the effect of hepatic impairment.
Preclinical Safety Data/Nonclinical Toxicology: Carcinogenicity: In a carcinogenicity study, doses up to 50 mg/kg/week were administered by subcutaneous injection to Sprague-Dawley male and female rats from 8 weeks to up to 98 weeks of age. These doses resulted in systemic exposures that were up to 19 times higher than the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg romosozumab (based on AUC comparison).
Romosozumab caused a dose-dependent increase in bone mass with macroscopic bone thickening at all doses. There were no effects of romosozumab on mortality or tumour incidence in male or female rats.
Mutagenicity: Mutagenesis has not been evaluated, as monoclonal antibodies are not expected to alter DNA or chromosomes.
Impairment of Fertility: No effects on fertility were observed in male and female rats at doses up to 300 mg/kg (100 times the clinical dose). No effects were noted in reproductive organs in rats and cynomolgus monkeys dosed with romosozumab in the 6-month chronic toxicology studies at exposures up to 37 and 90 times higher, respectively, than the systemic exposure observed in humans administered 210 mg romosozumab monthly (based on AUC comparison).
Animal Toxicology and/or Pharmacology: No adverse effects were noted in rats and monkeys after 26 once weekly subcutaneous injections at doses up to 100 mg/kg and systemic exposures 37 and 90 times higher, respectively, than the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg romosozumab (based on AUC comparison).
In growing rats administered a rodent surrogate sclerostin antibody at pharmacologically active doses, a transient increase in longitudinal growth rate predicted to result in < 1% increase in bone length was observed. In growing rats dosed with romosozumab for 6 months resulting in exposures up to 19 times higher than the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg romosozumab (based on AUC comparison), there was no effect on femur length.
In bone safety studies in ovariectomised rats and monkeys, once weekly treatment with romosozumab for 12 months increased bone formation and decreased bone resorption. The resulting increase in bone mass and improvements in cortical bone geometry and cancellous bone microarchitecture was associated with increased bone strength at exposures from 0.5 to 21 times higher than the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg romosozumab (based on AUC comparison).
Bone tissue was of normal or improved quality with no evidence of mineralisation defects, accumulation of osteoid, or woven bone.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image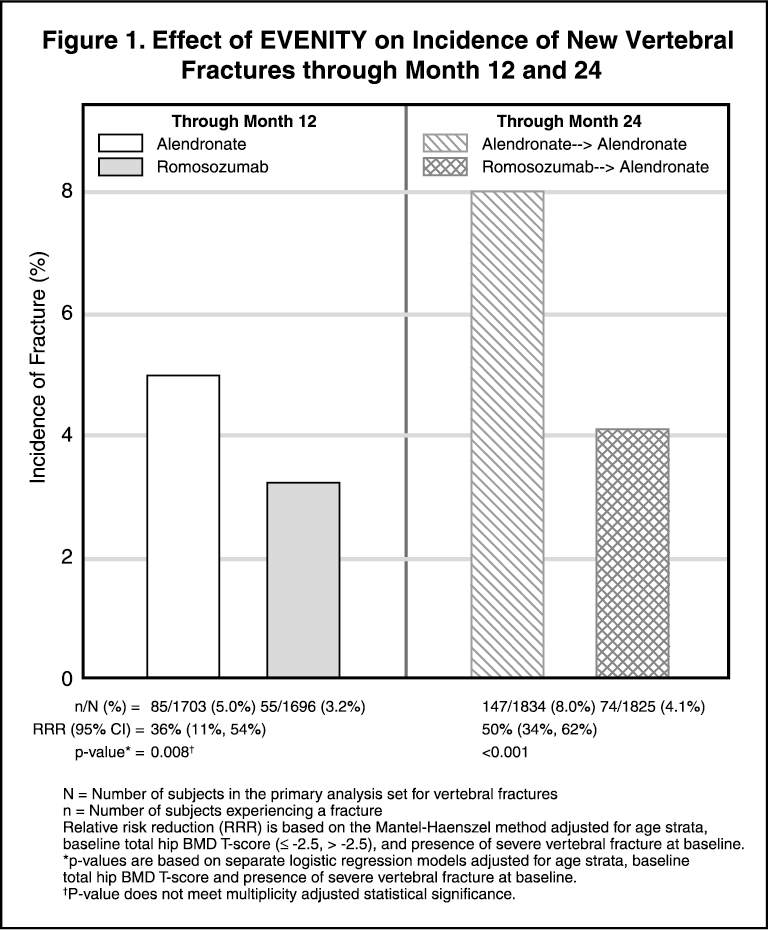 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image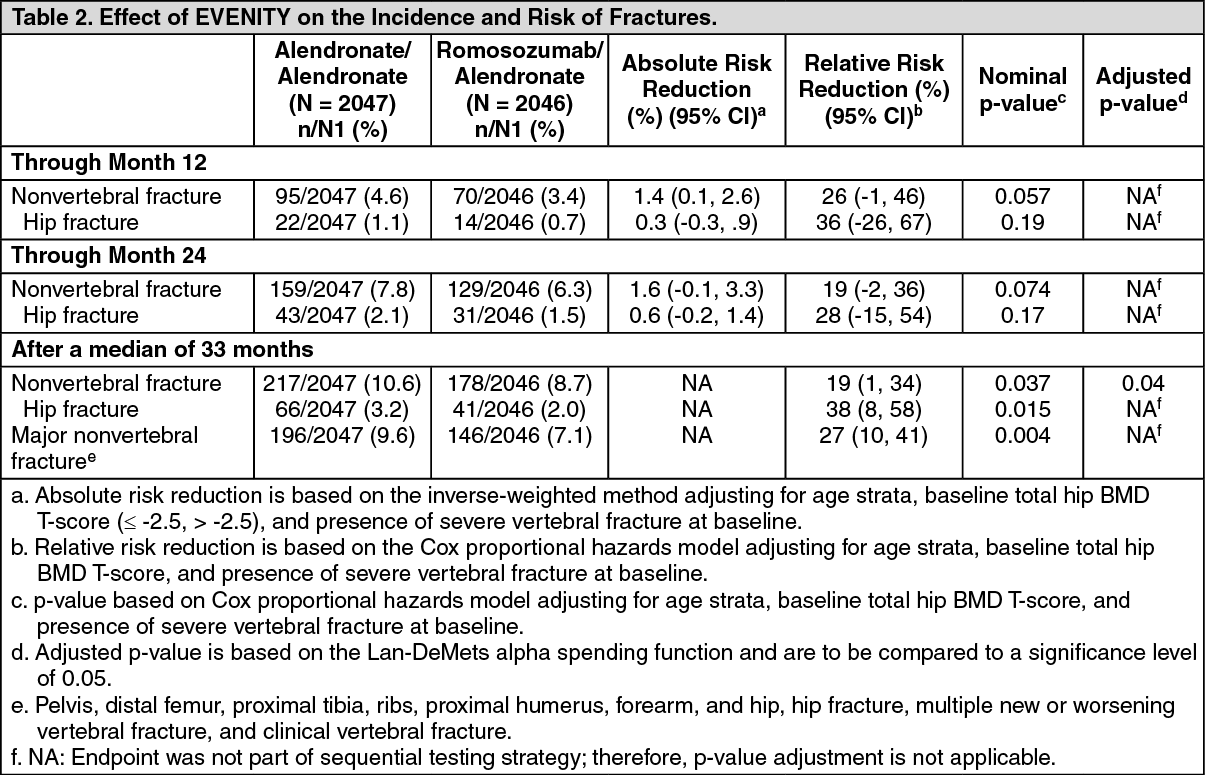 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image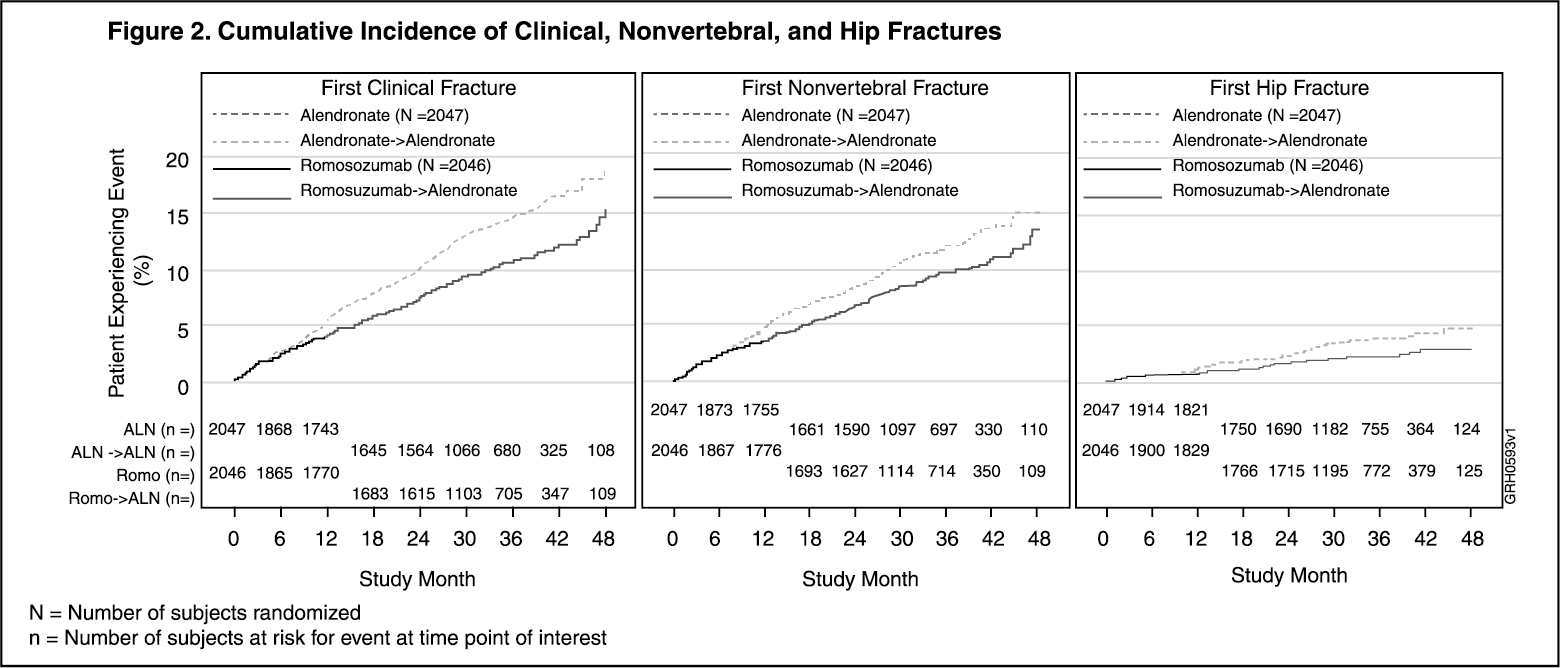 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image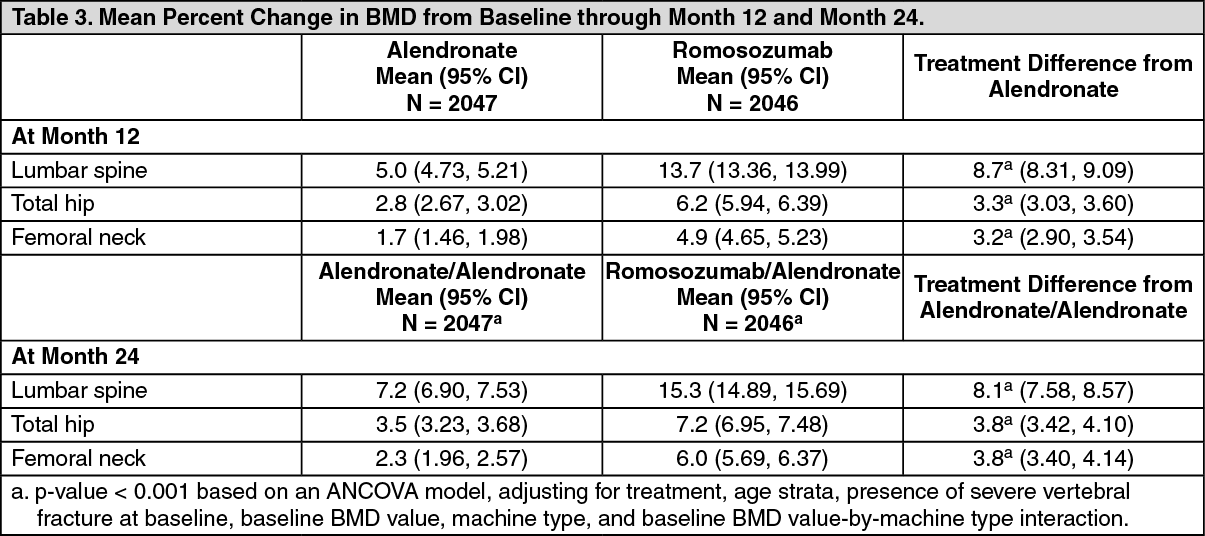 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image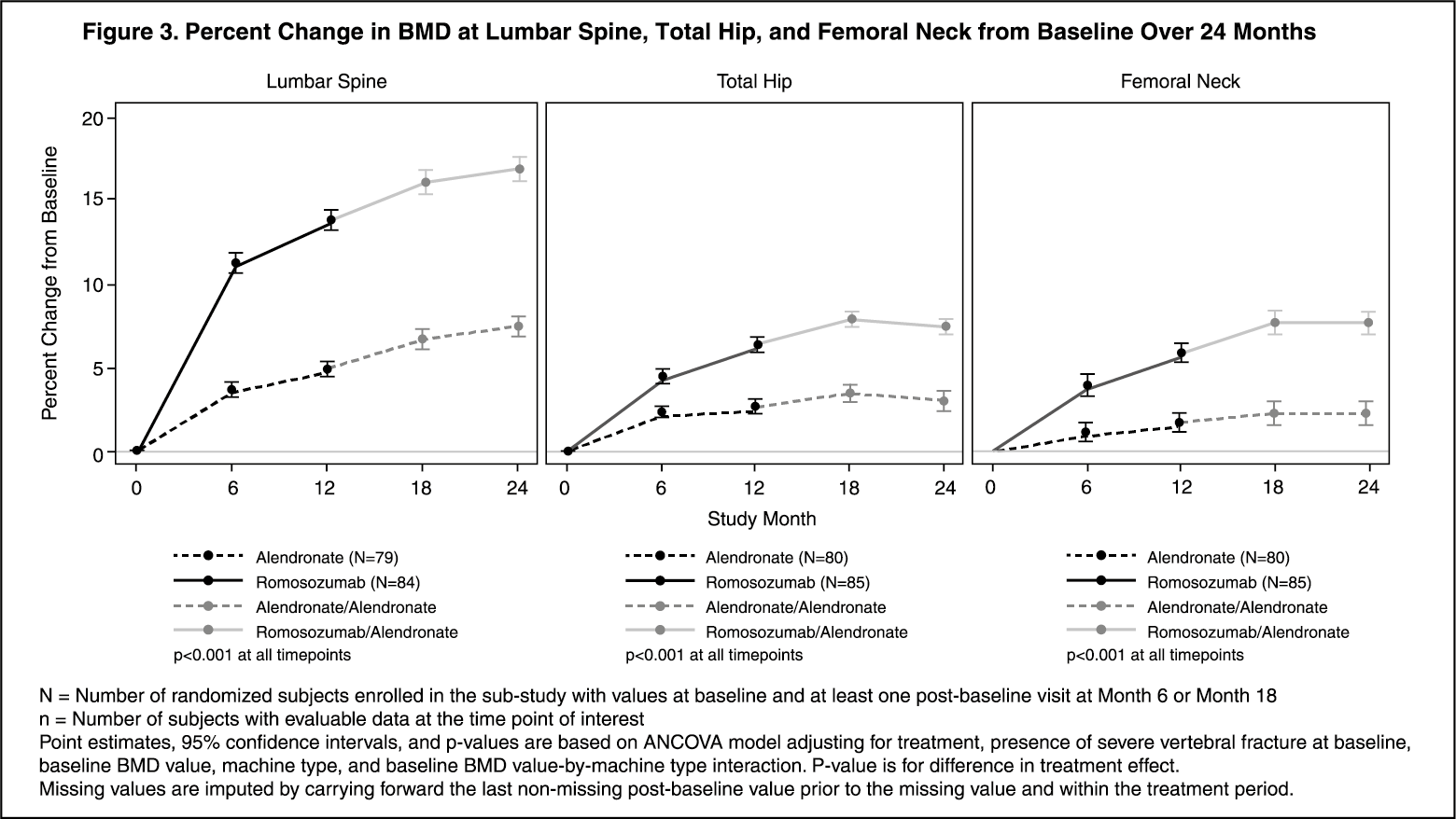 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image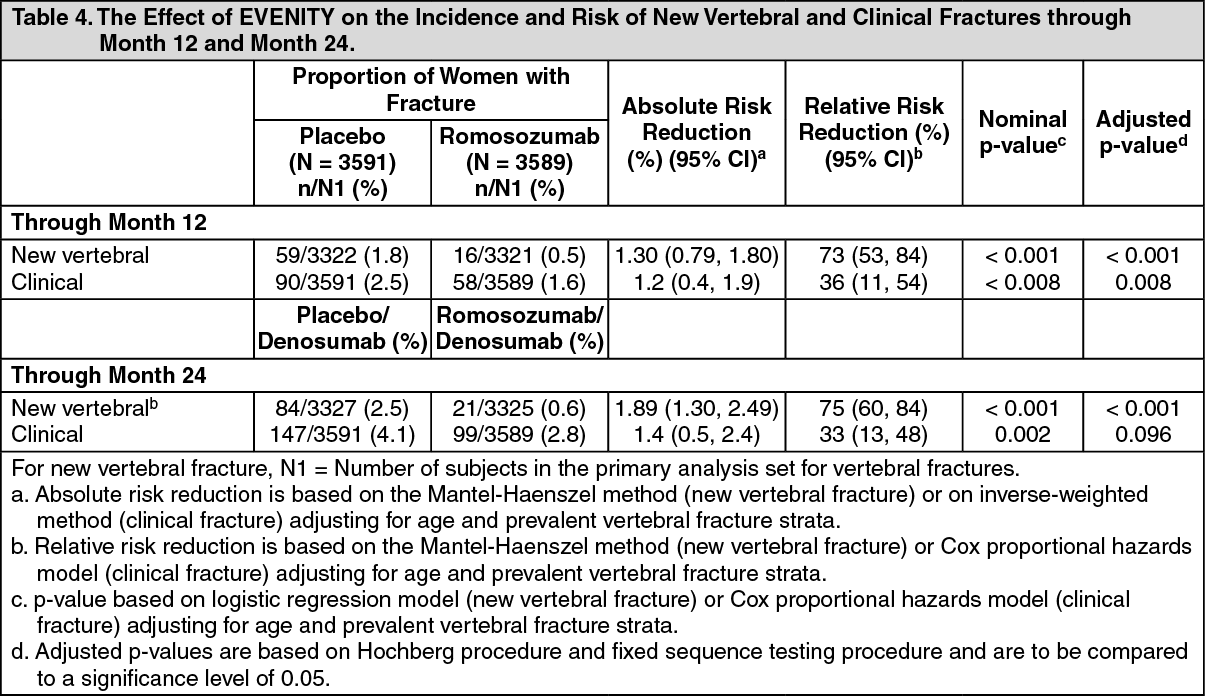 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image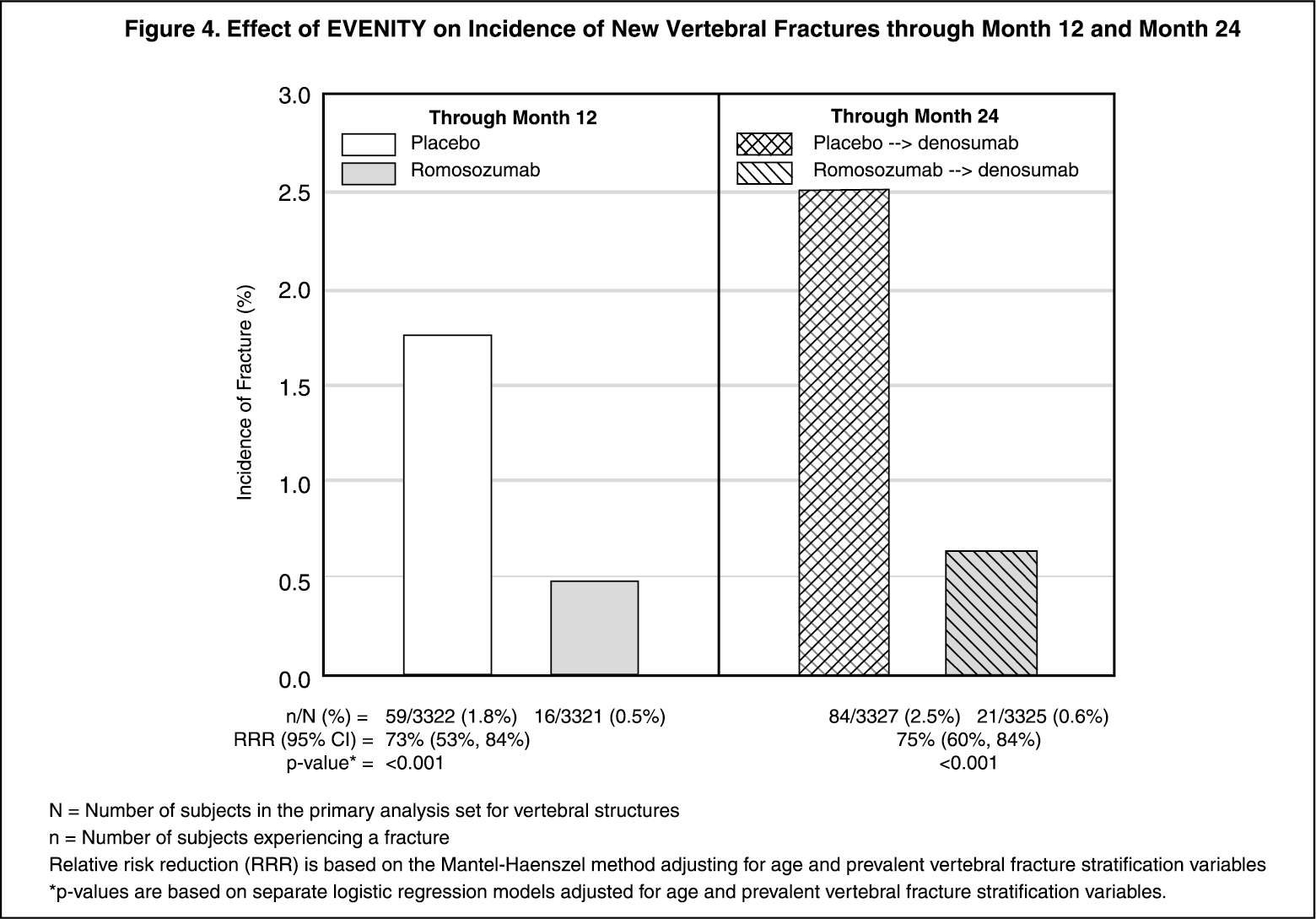 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image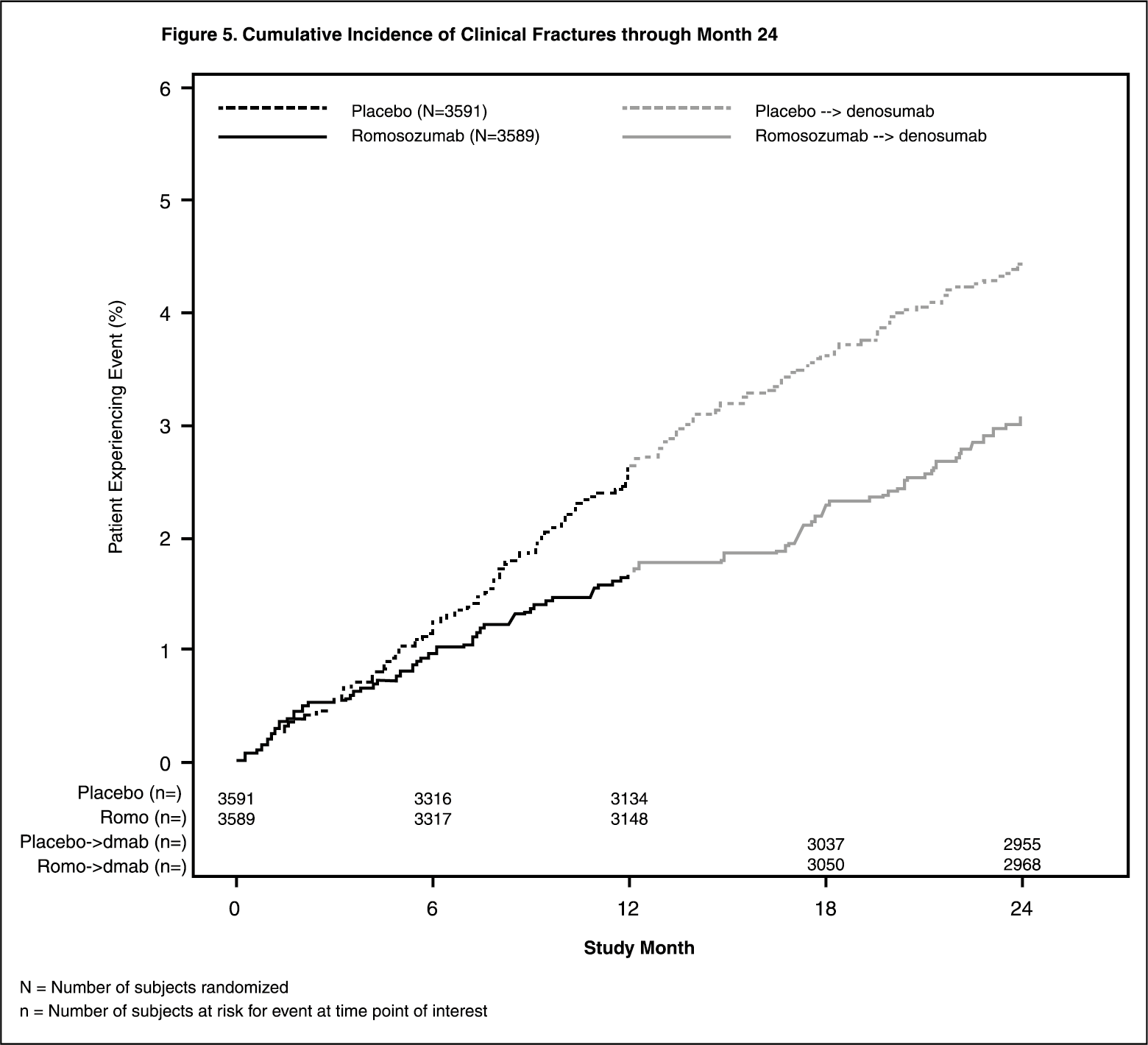 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image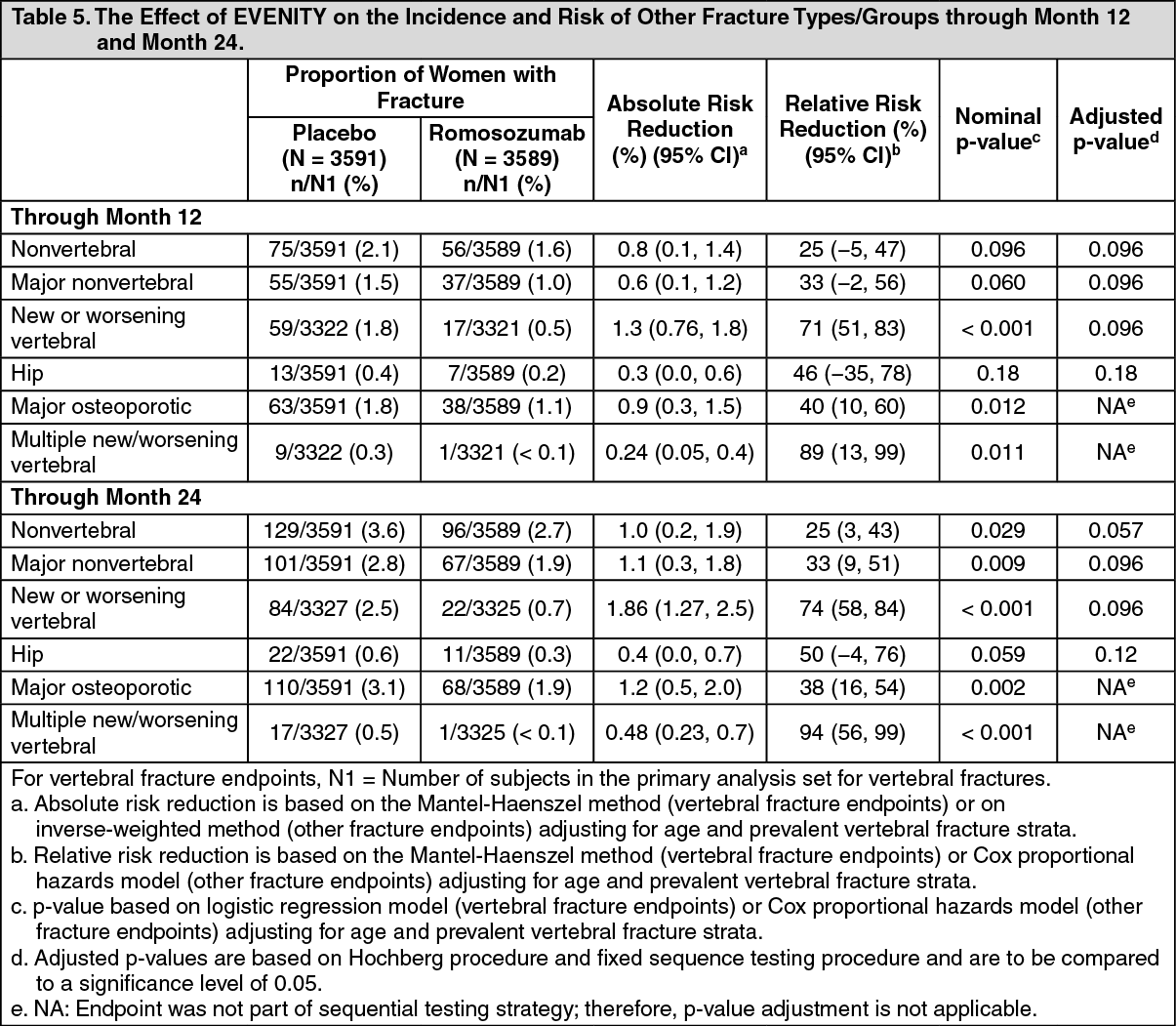 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image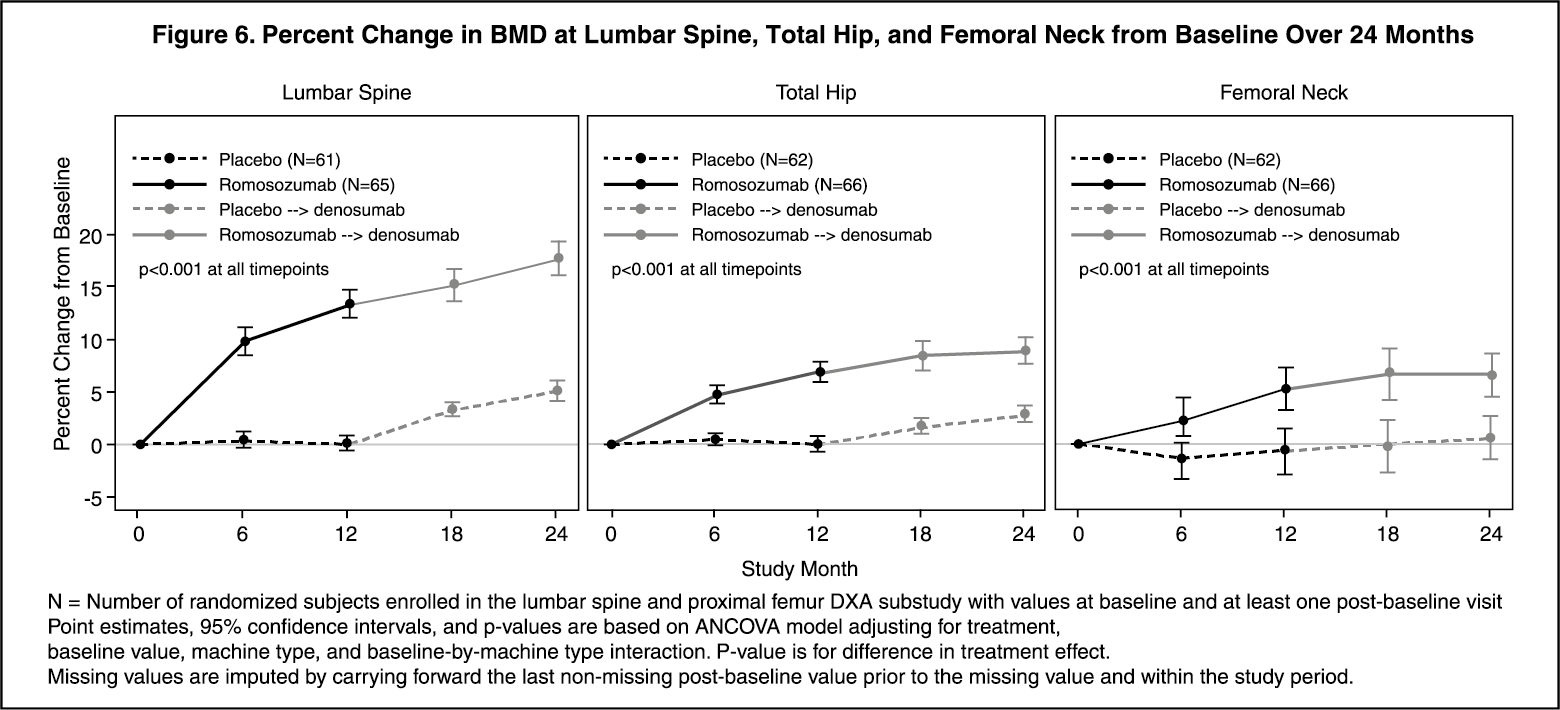 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image 105 mg_1.17 mLfe855b4b-b119-4286-9a9a-adb300bac0e1.GIF)

 Sign Out
Sign Out
