


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Mechanism of action: Apalutamide is an orally administered, selective Androgen Receptor (AR) inhibitor that binds directly to the ligand-binding domain of the AR. Apalutamide prevents AR nuclear translocation, inhibits DNA binding, impedes AR-mediated transcription, and lacks androgen receptor agonist activity. Apalutamide treatment decreases tumor cell proliferation and increases apoptosis leading to potent antitumor activity. A major metabolite, N-desmethyl apalutamide, exhibited one-third the in vitro activity of apalutamide.
Cardiac electrophysiology: The effect of apalutamide 240 mg once daily on the QTc interval was assessed in an open-label, uncontrolled, multi-center, single-arm dedicated QT study in 45 patients with CRPC. At steady-state, the maximum mean QTcF change from baseline was 12.4 ms (2-sided 90% upper CI: 16.0 ms). An exposure-QT analysis suggested a concentration-dependent increase in QTcF for apalutamide and its active metabolite.
Clinical efficacy and safety: The efficacy and safety of apalutamide has been established in two Phase 3 randomised, placebo-controlled studies, Study ARN-509-003 (nmCRPC) and 56021927PCR3002 (mHSPC).
TITAN: Metastatic Hormone-sensitive Prostate Cancer (mHSPC): TITAN was a randomised, double-blind, placebo-controlled, multinational, multicenter clinical trial in which 1052 patients with mHSPC were randomised (1:1) to receive either apalutamide orally at a dose of 240 mg once daily (N = 525) or placebo once daily (N = 527). All patients were required to have at least one bone metastasis on Technetium 99m bone scan. Patients were excluded if the site of metastases was limited to either the lymph nodes or viscera (e.g., liver or lung). All patients in the TITAN trial received concomitant GnRH analog or had prior bilateral orchiectomy. Around 11% of patients received prior treatment with docetaxel (maximum of 6 cycles, last dose ≤2 months prior to randomisation and maintained response prior to randomisation). The exclusion criteria included known brain metastases; prior treatment with other next generation anti-androgens (eg, enzalutamide), CYP17 inhibitors (eg, abiraterone acetate), immunotherapy (eg, sipuleucel-T), radiopharmaceutical agents or other treatments for prostate cancer; or history of seizure or condition that may predispose to seizure. Patients were stratified by Gleason score at diagnosis, prior docetaxel use, and region of the world. Patients with both high- and low-volume mHSPC were eligible for the study. High-volume disease was defined as either visceral metastases and at least 1 bone lesion or at least 4 bone lesions, with at least 1 bone lesion outside of the vertebral column or pelvis. Low-volume disease was defined as the presence of bone lesion(s) not meeting the definition of high-volume.
The following patient demographics and baseline disease characteristics were balanced between the treatment arms. The median age was 68 years (range 43-94) and 23% of patients were 75 years of age or older. The racial distribution was 68% Caucasian, 22% Asian, and 2% Black. Sixty-three percent (63%) of patients had high-volume disease and 37% had low-volume disease. Sixteen percent (16%) of patients had prior surgery, radiotherapy of the prostate or both. A majority of patients had a Gleason score of 7 or higher (92%). Sixty-eight percent (68%) of patients received prior treatment with a first generation anti-androgen in the non-metastatic setting. Although criteria for castration resistance were not determined at baseline, 94% of patients demonstrated a decrease in prostate specific antigen (PSA) from initiation of androgen deprivation therapy (ADT) to first dose of apalutamide or placebo. All patients except one in the placebo group, had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0 or 1 at study entry. Among the patients who discontinued study treatment (N = 271 for placebo and N = 170 for Erleada), the most common reason for discontinuation in both arms was disease progression. A greater proportion (73%) of patients treated with placebo received subsequent anti-cancer therapy compared to patients treated with Erleada (54%).
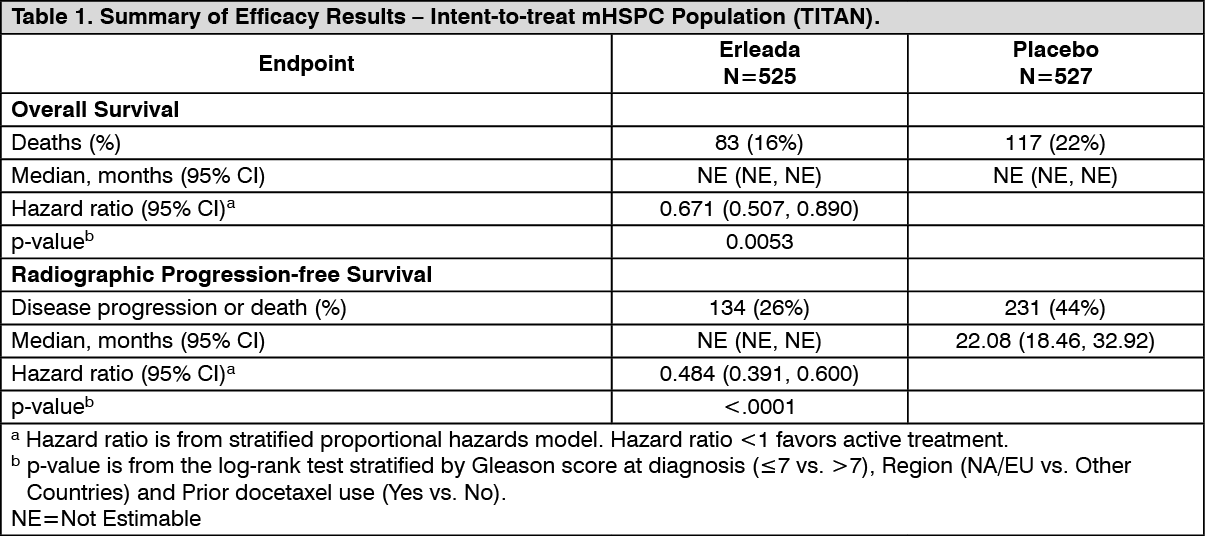
The major efficacy outcome measures of the study were overall survival (OS) and radiographic progression-free survival (rPFS). Efficacy results of TITAN are summarised in Table 1 and Figures 1 and 2. (See Table 1.)
 Click on icon to see table/diagram/image
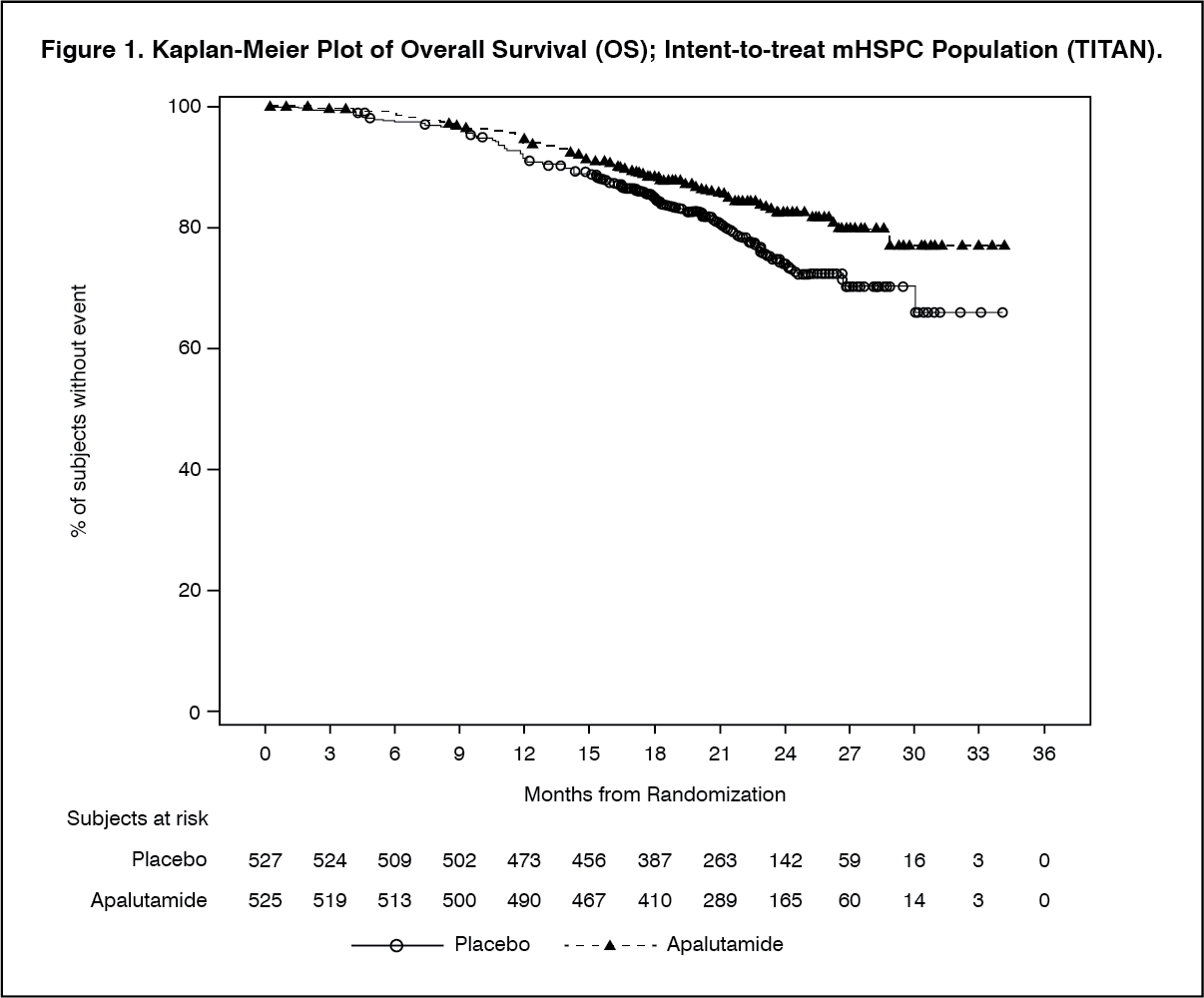
Click on icon to see table/diagram/imageA statistically significant improvement in OS and rPFS was demonstrated in patients randomised to receive Erleada compared with patients randomised to receive placebo. Consistent improvement in rPFS was observed across patient subgroups including high- or low-volume disease, prior docetaxel use (yes or no), age (< 65, ≥65, or ≥75 years old), baseline PSA above median (yes or no), and number of bone lesions (≤10 or >10). (See Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image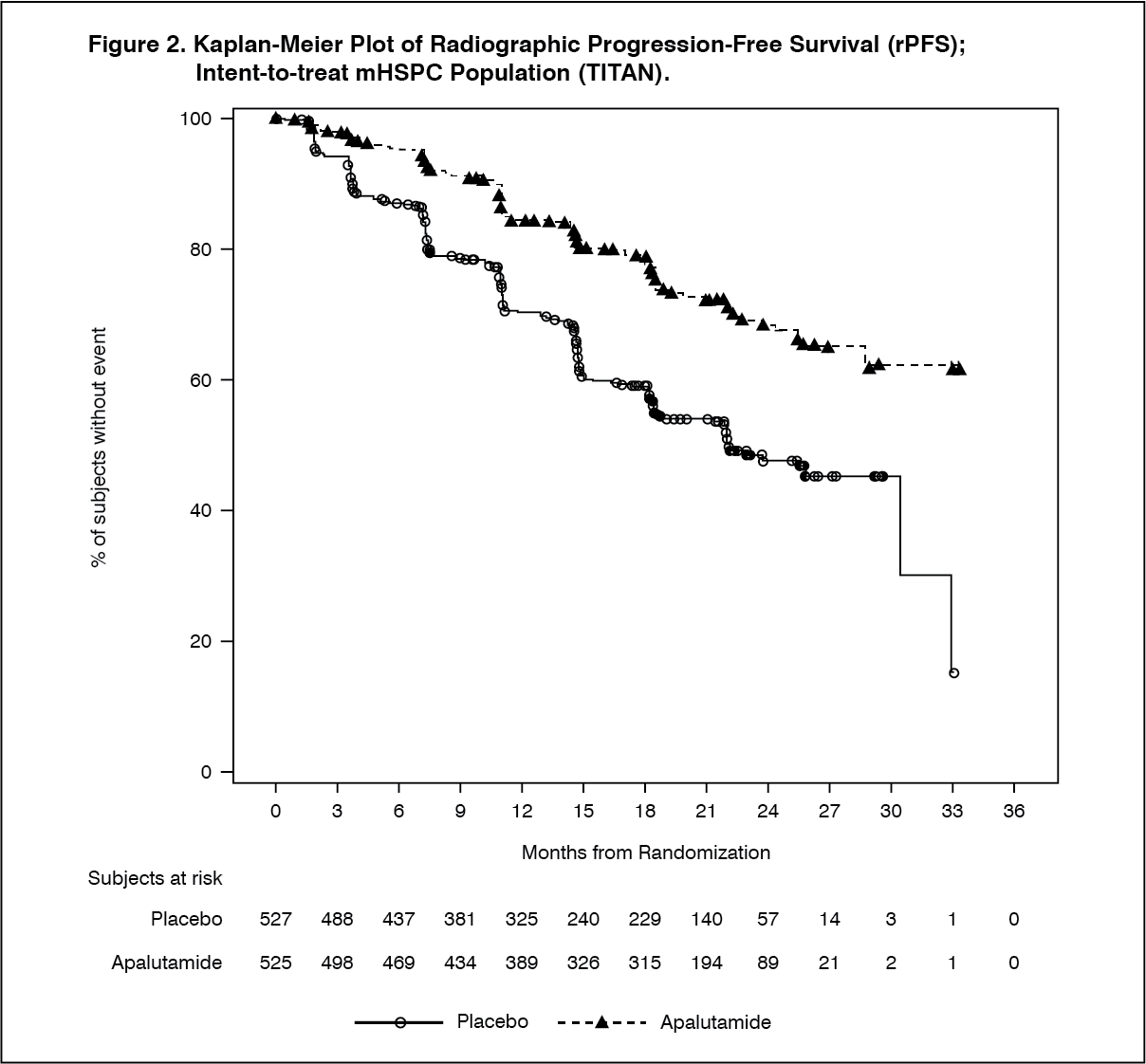 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment with Erleada statistically significantly delayed the initiation of cytotoxic chemotherapy (HR = 0.391, CI = 0.274, 0.558; p < 0.0001), resulting in a 61% reduction of risk for subjects in the treatment arm compared to the placebo arm.
SPARTAN: Non-Metastatic Castration Resistant Prostate Cancer (nmCRPC): A total of 1207 subjects with NM-CRPC were randomised 2:1 to receive either apalutamide orally at a dose of 240 mg once daily in combination with androgen deprivation therapy (ADT) (medical castration or prior surgical castration) or placebo with ADT in a multicenter, double-blind, clinical study (Study ARN-509-003). Subjects enrolled had a Prostate Specific Antigen (PSA) Doubling Time (PSADT) ≤ 10 months, considered to be at high risk of imminent metastatic disease and prostate cancer-specific death. All subjects who were not surgically castrated received ADT continuously throughout the study. PSA results were blinded and were not used for treatment discontinuation. Subjects randomised to either arm were to continue treatment until disease progression defined by blinded central imaging review (BICR), initiation of new treatment, unacceptable toxicity or withdrawal.
The following patient demographics and baseline disease characteristics were balanced between the treatment arms. The median age was 74 years (range 48-97) and 26% of subjects were 80 years of age or older. The racial distribution was 66% Caucasian, 5.6% Black, 12% Asian, and 0.2% Other. Seventy-seven percent (77%) of subjects in both treatment arms had prior surgery or radiotherapy of the prostate. A majority of subjects had a Gleason score of 7 or higher (81%). Fifteen percent (15%) of subjects had < 2 cm pelvic lymph nodes at study entry. Seventy-three percent (73%) of subjects received prior treatment with a first generation anti-androgen; 69% of subjects received bicalutamide and 10% of subjects received flutamide. All subjects enrolled were confirmed to be non-metastatic by blinded central imaging review and had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) performance status score of 0 or 1 at study entry.
Metastasis-free survival (MFS) was the primary endpoint, defined as the time from randomisation to the time of first evidence of BICR-confirmed bone or soft tissue distant metastasis or death due to any cause, whichever occurred first. Treatment with ERLEADA significantly improved MFS. ERLEADA decreased the relative risk of distant metastasis or death by 70% compared to placebo (HR = 0.30; 95% CI: 0.24, 0.36; p < 0.0001). The median MFS for ERLEADA was 41 months and was 16 months for placebo (see Figure 3). Consistent improvement in MFS with ERLEADA was observed for all pre-specified subgroups, including age, race, region of the world, nodal status, prior number of hormonal therapies, baseline PSA, PSA doubling time, baseline ECOG status and use of bone-sparing agents. (See Figure 3.)
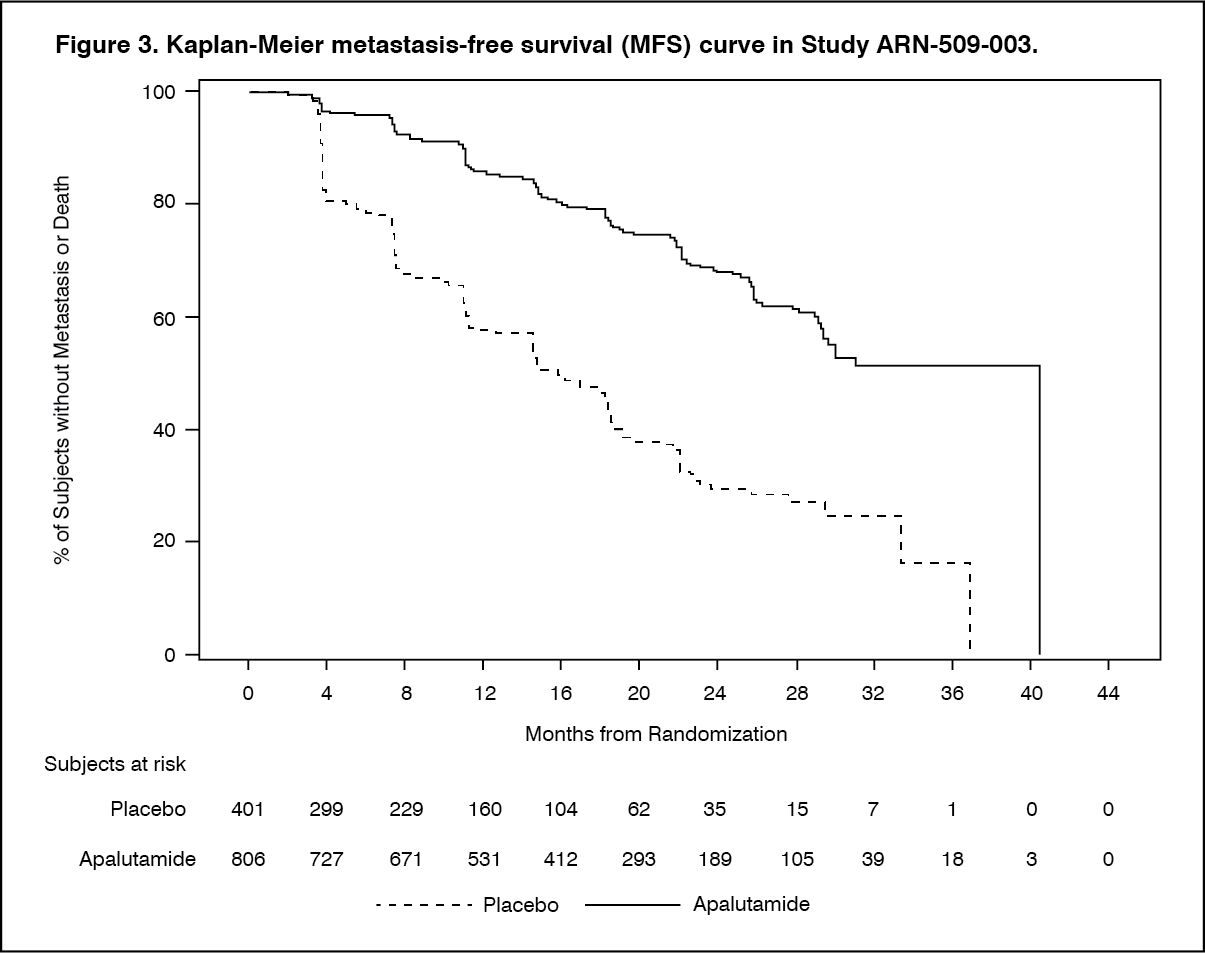 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTaking account of all data, subjects treated with ERLEADA and ADT showed significant improvement over those treated with ADT alone for the following secondary endpoints of time to metastasis (HR = 0.28; 95% CI: 0.23, 0.34; p < 0.0001), progression-free survival (PFS) (HR = 0.30; 95% CI: 0.25, 0.36; p < 0.0001); time to symptomatic progression (HR = 0.57; 95% CI: 0.44, 0.73; p < 0.0001); overall survival (OS) (HR = 0.78; 95% CI: 0.64, 0.96; p = 0.0161) and time to initiation of cytotoxic chemotherapy (HR = 0.63; 95% CI: 0.49, 0.81; p = 0.0002).
Time to symptomatic progression was defined as time from randomisation to development of a skeletal related event, pain/symptoms requiring initiation of a new systemic anti-cancer therapy, or loco-regional tumor progression requiring radiation/surgery. While the overall number of events was small, the difference between the two arms was sufficiently large to reach statistical significance. Treatment with ERLEADA decreased the risk of symptomatic progression by 43% compared with placebo (HR=0.567; 95% CI: 0.443, 0.725; p < 0.0001. The median time to symptomatic progression was not reached in either treatment group.
With median follow-up time of 52.0 months, results showed that treatment with ERLEADA significantly decreased the risk of death by 22% compared with placebo (HR = 0.784; 95% CI: 0.643, 0.956; 2 sided p = 0.0161). The median OS was 73.9 months for the ERLEADA arm and 59.9 months for the placebo arm. The pre-specified alpha boundary (p ≤ 0.046) was crossed and statistical significance was achieved. This improvement was demonstrated even though 19% of patients in the placebo arm received ERLEADA as subsequent therapy. (See Figure 4.)
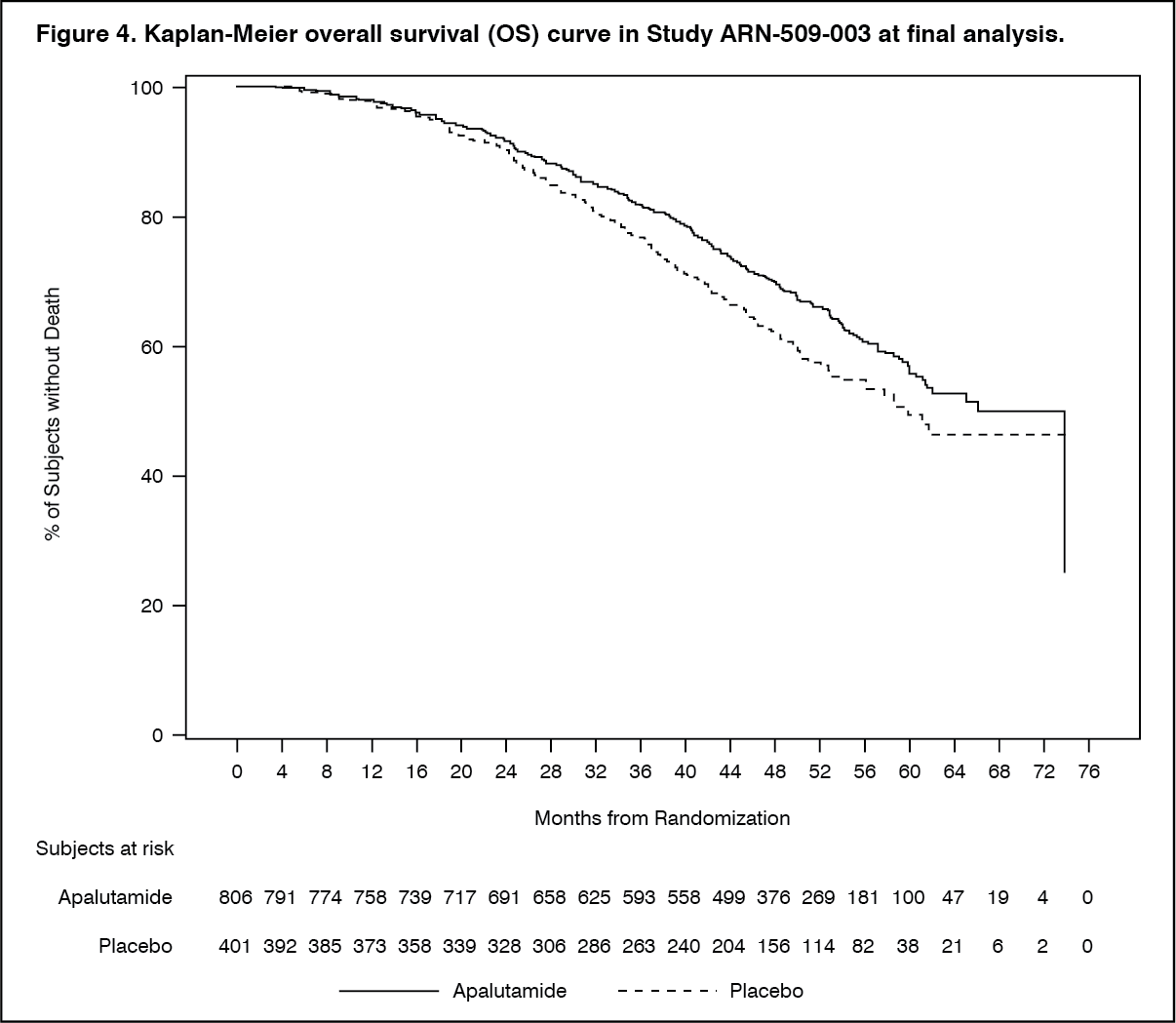 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment with ERLEADA significantly decreased the risk of initiating cytotoxic chemotherapy by 37% compared with placebo (HR = 0.629; 95% CI: 0.489, 0.808; p = 0.0002) demonstrating statistically significant improvement for ERLEADA versus placebo. The median time to the initiation of cytotoxic chemotherapy was not reached for either treatment arm.
PFS 2, defined as the time to death or disease progression by PSA, radiographic, or symptomatic progression on or after first subsequent therapy was longer for subjects treated with ERLEADA compared to those treated with placebo. Results demonstrated a 44% reduction in risk of PFS-2 with ERLEADA versus placebo (HR = 0.565, 95% CI: 0.471, 0.677; p < 0.0001).
There were no detrimental effects to overall health-related quality of life with the addition of ERLEADA to ADT and a small but not clinically meaningful difference in change from baseline in favor of ERLEADA observed in the analysis of the Functional Assessment of Cancer Therapy Prostate (FACT P) total score and subscales.
Pharmacokinetics: Following repeat once-daily dosing, apalutamide exposure (Cmax and area under the concentration curve [AUC]) increased in a dose-proportional manner across the dose range of 30 to 480 mg. Following administration of 240 mg once daily, apalutamide steady state was achieved after 4 weeks and the mean accumulation ratio was approximately 5-fold relative to a single dose. At steady-state, mean (CV%) Cmax and AUC values for apalutamide were 6 μg/mL (28%) and 100 μg.h/mL (32%), respectively. Daily fluctuations in apalutamide plasma concentrations were low, with mean peak-to-trough ratio of 1.63. An increase in apparent clearance (CL/F) was observed with repeat dosing, likely due to induction of apalutamide's own metabolism.
At steady-state, the mean (CV%) Cmax and AUC values for the major active metabolite, N-desmethyl apalutamide, were 5.9 μg/mL (18%) and 124 μg.h/mL (19%), respectively. N-desmethyl apalutamide is characterised by a flat concentration-time profile at steady-state with a mean peak-to-trough ratio of 1.27. Mean (CV%) AUC metabolite/parent drug ratio for N-desmethyl apalutamide following repeat-dose administration was about 1.3 (21%). Based on systemic exposure, relative potency, and pharmacokinetic properties, N-desmethyl apalutamide likely contributed to the clinical activity of apalutamide.
Absorption: After oral administration, median time to achieve peak plasma concentration (tmax) was 2 hours (range: 1 to 5 hours). Mean absolute oral bioavailability is approximately 100%, indicating that apalutamide is completely absorbed after oral administration.
Administration of apalutamide to healthy subjects under fasting conditions and with a high-fat meal resulted in no clinically relevant changes in Cmax and AUC. Median time to reach tmax was delayed about 2 hours with food (see Dosage & Administration).
Apalutamide is not ionizable under relevant physiological pH condition, therefore acid lowering agents (e.g., proton pump inhibitor, H2-receptor antagonist, antacid) are not expected to affect the solubility and bioavailability of apalutamide.
In vitro, apalutamide and its N-desmethyl metabolite are substrates for P-gp. Because apalutamide is completely absorbed after oral administration, P-gp does not limit the absorption of apalutamide and therefore, inhibition or induction of P-gp is not expected to affect the bioavailability of apalutamide.
Distribution: The mean apparent volume of distribution at steady-state of apalutamide is about 276 L. The volume of distribution of apalutamide is greater than the volume of total body water, indicative of extensive extravascular distribution.
Apalutamide and N-desmethyl apalutamide are 96% and 95% bound to plasma proteins, respectively, and mainly bind to serum albumin with no concentration dependency.
Biotransformation: Following single oral administration of 14C-labeled apalutamide 240 mg, apalutamide, the active metabolite, N-desmethyl apalutamide, and an inactive carboxylic acid metabolite accounted for the majority of the 14C-radioactivity in plasma, representing 45%, 44%, and 3%, respectively, of the total 14C-AUC.
Metabolism is the main route of elimination of apalutamide. It is metabolised primarily by CYP2C8 and CYP3A4 to form N-desmethyl apalutamide. Apalutamide and N-desmethyl apalutamide are further metabolised to form the inactive carboxylic acid metabolite by carboxylesterase. The contribution of CYP2C8 and CYP3A4 in the metabolism of apalutamide is estimated to be 58% and 13% following single dose but the level of contribution is expected to change at steady-state due to induction of CYP3A4 by apalutamide after repeat dose.
Elimination: Apalutamide, mainly in the form of metabolites, is eliminated primarily via urine. Following a single oral administration of radiolabeled apalutamide, 89% of the radioactivity was recovered up to 70 days post-dose: 65% was recovered in urine (1.2% of dose as unchanged apalutamide and 2.7% as N-desmethyl apalutamide) and 24% was recovered in feces (1.5% of dose as unchanged apalutamide and 2% as N-desmethyl apalutamide).
The apparent oral clearance (CL/F) of apalutamide is 1.3 L/h after single dosing and increases to 2.0 L/h at steady-state after once-daily dosing. The mean effective half-life for apalutamide in patients is about 3 days at steady-state.
In vitro data indicate that apalutamide and its N-desmethyl metabolite are not substrates for BCRP, OATP1B1 or OATP1B3.
Special populations: The effects of renal impairment, hepatic impairment, age, race, and other extrinsic factors on the pharmacokinetics of apalutamide are summarised as follows.
Renal impairment: A dedicated renal impairment study for apalutamide has not been conducted. Based on the population pharmacokinetic analysis using data from clinical studies in subjects with castration-resistant prostate cancer (CRPC) and healthy subjects, no significant difference in systemic apalutamide exposure was observed in subjects with pre-existing mild to moderate renal impairment (estimated glomerular filtration rate [eGFR] between 30 to 89 mL/min/1.73 m2; N=585) compared to subjects with baseline normal renal function (eGFR ≥ 90 mL/min/1.73 m2; N=372). The potential effect of severe renal impairment or end stage renal disease (eGFR ≤29 mL/min/1.73 m2) have not been established due to insufficient data.
Hepatic impairment: A dedicated hepatic impairment study compared the systemic exposure of apalutamide and N- desmethyl apalutamide in subjects with baseline mild hepatic impairment (N=8, Child-Pugh Class A, mean score = 5.3) or moderate hepatic impairment (N=8, Child-Pugh Class B, mean score = 7.6) versus healthy controls with normal hepatic function (N=8). Following a single oral 240 mg dose of apalutamide, the geometric mean ratio (GMR) for AUC and Cmax for apalutamide in subjects with mild impairment was 95% and 102%, respectively, and the GMR for AUC and Cmax of apalutamide in subjects with moderate impairment was 113% and 104%, respectively, compared to healthy control subjects. Clinical and pharmacokinetic data for apalutamide are not available for patients with severe hepatic impairment (Child-Pugh Class C).
Ethnicity and race: Based on population pharmacokinetic analysis, there were no clinically relevant differences in apalutamide pharmacokinetics between White (Caucasian or Hispanic or Latino; N=761), Black (of African heritage or African American; N=71), Asian (non-Japanese; N=58) and Japanese (N=58).
Age: Population pharmacokinetic analyses showed that age (range: 18 to 94 years) does not have a clinically meaningful influence on the pharmacokinetics of apalutamide.
Toxicology: Preclinical safety data: Apalutamide was negative for genotoxicity in a standard battery of in vitro and in vivo tests.
Apalutamide was not carcinogenic in a 6-month study in the male transgenic (Tg.rasH2) mouse at doses up to 30 mg/kg per day, which is 1.2 and 0.5 times for apalutamide and N-desmethyl apalutamide respectively, the clinical exposure (AUC) at the recommended clinical dose of 240 mg/day.
Male fertility is likely to be impaired by treatment with apalutamide based on findings in repeat-dose toxicology studies which were consistent with the pharmacological activity of apalutamide. In repeat-dose toxicity studies in male rats and dogs, atrophy, aspermia/hypospermia, degeneration and/or hyperplasia or hypertrophy in the reproductive system were observed at doses corresponding to exposures approximately equal to the human exposure based on AUC.
In a fertility study in male rats, a decrease in sperm concentration and motility, copulation and fertility rates (upon pairing with untreated females) along with reduced weights of the secondary sex glands and epididymis were observed following 4 weeks of dosing at doses corresponding to exposures approximately equal to the human exposure based on AUC. Effects on male rats were reversible after 8 weeks from the last apalutamide administration.
In a preliminary embryofetal developmental toxicity study in rats, apalutamide caused developmental toxicity when administered at oral doses of 25, 50 or 100 mg/kg/day throughout the period of organogenesis (gestational days 6-20). These doses resulted in systemic exposures approximately 2, 4 and 6 times, respectively, on an AUC basis, the exposure in humans at the dose of 240 mg/day. Findings included non pregnant females at 100 mg/kg/day and embryofetal lethality (resorptions) at doses ≥50 mg/kg/day, decreased fetal anogenital distance and a misshapen pituitary gland (more rounded shape) at ≥25 mg/kg/day. Skeletal variations (unossified phalanges, supernumerary short thoracolumbar rib(s) and/or abnormalities of the hyoid) were also noted at doses ≥25 mg/kg/day, without resulting in an effect on mean fetal weight.