


 Sign Out
Sign Out

Infusion-related reactions were reported in 4% of patients receiving Vedolizumab.
In the shorter (10-week) placebo controlled induction trial the types of adverse reactions reported were similar but occurred at lower frequency than the longer 52-Week trials. A further 279 patients were treated with Vedolizumab at Week 0 and Week 2 and then with placebo for up to 52 weeks. Of these patients, 84% experienced adverse events and 15% experienced serious adverse events. Patients (n=1822) previously enrolled in Phase 2 or 3 Vedolizumab studies were eligible to enroll in an ongoing open-label study and received Vedolizumab 300 mg every four weeks.
The following convention is used for the classification of the frequency of an adverse drug reaction (ADR) and is based on the Council for International Organizations of Medical Sciences (CIOMS) guidelines: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to< 1/1,000); very rare (< 1/10,000); not known (cannot be estimated from the available data). (See Table 5.)
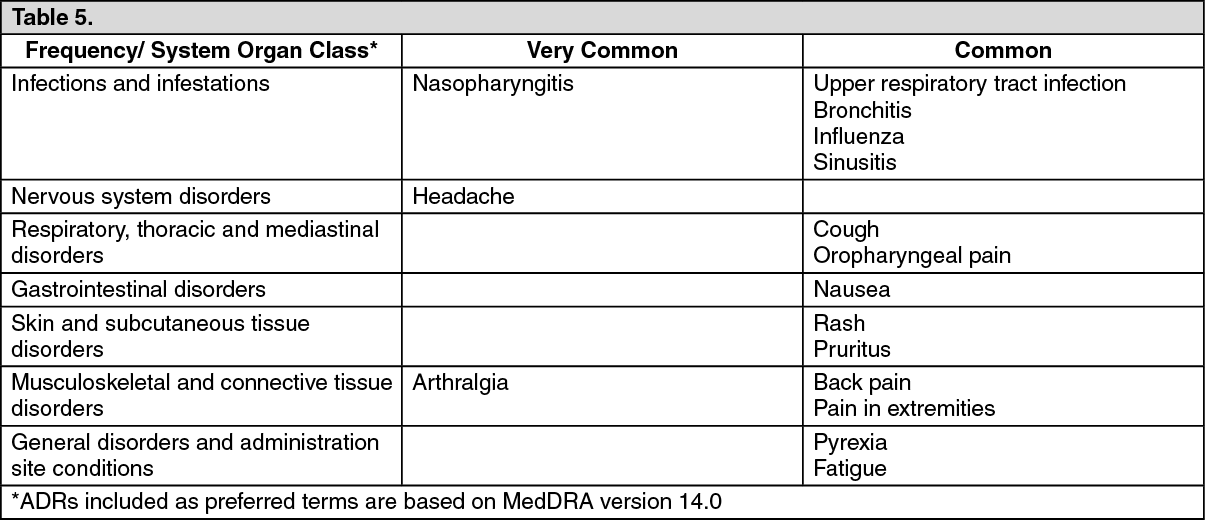 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePostmarketing Experience: In the post-marketing setting reports of anaphylaxis have been identified. The frequency of anaphylaxis in this setting is unknown.
Description of selected adverse reactions: Infusion-related reactions: In the 52-Week controlled studies, 4% of Vedolizumab-treated patients and 3% of placebo-treated patients experienced an adverse event defined by the investigator as IRR. The majority of IRRs were mild or moderate in intensity and <1% resulted in discontinuation of study treatment. Observed IRRs generally resolved with no or minimal intervention following the infusion. Most infusion related reactions occurred within the first 2 hours. Of those patients who had IRRs, those dosed with Vedolizumab had more IRRs within the first two hours as compared to placebo patients with IRRs. Most IRRs were not serious and occurred during the infusion or within the first hour after infusion is completed.
One serious adverse event of IRR was reported in a Crohn's disease patient during the second infusion (symptoms reported were dyspnea, bronchospasm, urticaria, flushing, rash, and increased blood pressure and heart rate) and was successfully managed with discontinuation of infusion and treatment with antihistamine and intravenous hydrocortisone. In patients who received Vedolizumab at Weeks 0 and 2 followed by placebo, no increase in the rate of IRR was seen upon retreatment with Vedolizumab after loss of response.
Infections: In the 52-Week controlled studies, the rate of infections was 0.85 per patient-year in the Vedolizumab-treated patients and 0.70 per patient-year in the placebo-treated patients. The infections consisted primarily of nasopharyngitis, upper respiratory tract infection, sinusitis, and urinary tract infections. Most patients continued on Vedolizumab after the infection resolved.
In the 52-Week controlled studies, the rate of serious infections was 0.07 per patient year in Vedolizumab-treated patients and 0.06 per patient year in placebo-treated patients. Over time, there was no significant increase in the rate of serious infections.
In controlled and open-label studies in adults treated with Vedolizumab, serious infections have been reported, which include tuberculosis, sepsis (some fatal), salmonella sepsis, Listeria meningitis, and cytomegaloviral colitis.
In clinical studies with intravenous vedolizumab, the rate of infections in vedolizumab treated patients with BMI of 30 kg/m2 and above was higher than for those with BMI less than 30 kg/m2.
Immunogenicity: An acid dissociation electrochemiluminescence (ECL) method for detecting antibodies to vedolizumab was developed and validated. The incidence of anti-vedolizumab antibodies to intravenous vedolizumab with the drug-tolerant ECL method for patients in GEMINI 1 and GEMINI 2 studies who had continuous treatment for 52 weeks was 6% (86 out of 1427). Of the 86 patients who tested positive for anti-vedolizumab antibodies, 20 patients were persistently positive and 56 developed neutralizing antibodies to vedolizumab. Overall, there was no apparent correlation of anti-vedolizumab antibody development to adverse events following administration of vedolizumab.
Malignancy: Overall, results from the clinical program to date do not suggest an increased risk for malignancy with Vedolizumab treatment; however, the number of malignancies was small and long-term exposure was limited.
Long-term safety evaluations are ongoing.
View ADR Monitoring Form