Pharmacotherapeutic group: Agents acting on the renin-angiotensin system; angiotensin II receptor blockers (ARBs), other combinations.
ATC code: C09DX04.
Pharmacology: Pharmacodynamics: Mechanism of action: Sacubitril/valsartan exhibits the mechanism of action of an angiotensin receptor neprilysin inhibitor by simultaneously inhibiting neprilysin (neutral endopeptidase; NEP) via LBQ657, the active metabolite of the prodrug sacubitril, and by blocking the angiotensin II type-1 (AT1) receptor via valsartan. The complementary cardiovascular benefits of sacubitril/valsartan in heart failure patients are attributed to the enhancement of peptides that are degraded by neprilysin, such as natriuretic peptides (NP), by LBQ657 and the simultaneous inhibition of the effects of angiotensin II by valsartan. NPs exert their effects by activating membrane-bound guanylyl cyclase-coupled receptors, resulting in increased concentrations of the second messenger cyclic guanosine monophosphate (cGMP), which could result in vasodilation, natriuresis and diuresis, increased glomerular filtration rate and renal blood flow, inhibition of renin and aldosterone release, reduction of sympathetic activity, and anti-hypertrophic and anti-fibrotic effects.
Valsartan inhibits detrimental cardiovascular and renal effects of angiotensin II by selectively blocking the AT1 receptor, and also inhibits angiotensin II-dependent aldosterone release. This prevents sustained activation of the renin-angiotensin-aldosterone system that would result in vasoconstriction, renal sodium and fluid retention, activation of cellular growth and proliferation, and subsequent maladaptive cardiovascular remodelling.
Pharmacodynamic effects: The pharmacodynamic effects of sacubitril/valsartan were evaluated after single and multiple dose administrations in healthy subjects and in patients with heart failure, and are consistent with simultaneous neprilysin inhibition and RAAS blockade. In a 7-day valsartan-controlled study in patients with reduced ejection fraction (HFrEF), administration of sacubitril/valsartan resulted in an initial increase in natriuresis, increased urine cGMP, and decreased plasma levels of mid-regional pro-atrial natriuretic peptide (MR-proANP) and N-terminal prohormone brain natriuretic peptide (NT-proBNP) compared to valsartan. In a 21-day study in HFrEF patients, sacubitril/valsartan significantly increased urine ANP and cGMP and plasma cGMP, and decreased plasma NT-proBNP, aldosterone and endothelin-1 compared to baseline. The AT1-receptor was also blocked as evidenced by increased plasma renin activity and plasma renin concentrations. In the PARADIGM-HF study, sacubitril/valsartan decreased plasma NT-proBNP and increased plasma BNP and urine cGMP compared with enalapril. BNP is not a suitable biomarker of heart failure in patients treated with sacubitril/valsartan because BNP is a neprilysin substrate (see Precautions). NT-proBNP is not a neprilysin substrate and is therefore a more suitable biomarker.
In PARAMOUNT, a randomized, double-blind, 36-week study in patients with heart failure with LVEF ≥45% comparing 200 mg of Entresto (n=149) to 160 mg of valsartan (n=152) twice-daily, Entresto decreased NT-proBNP by 17% while valsartan increased NT-proBNP by 8% at Week 12 (p=0.005).
In PARAGON-HF, Entresto decreased NT-proBNP by 24% (Week 16) and 19% (Week 48) compared to 6% and 3% reductions on valsartan, respectively.
In a thorough QTc clinical study in healthy male subjects, single doses of sacubitril/valsartan 194 mg sacubitril/206 mg valsartan and 583 mg sacubitril/617 mg valsartan had no effect on cardiac repolarisation.
Neprilysin is one of multiple enzymes involved in the clearance of amyloid-β (Aβ) from the brain and cerebrospinal fluid (CSF). Administration of sacubitril/valsartan 194 mg sacubitril/206 mg valsartan once daily for two weeks to healthy subjects was associated with an increase in CSF Aβ1-38 compared to placebo; there were no changes in concentrations of CSF Aβ1-40 and 1-42. The clinical relevance of this finding is not known (see Toxicology: Preclinical safety data as follows).
Clinical efficacy and safety: The 24 mg/26 mg, 49 mg/51 mg and 97 mg/103 mg strengths are in some publications referred to as 50, 100 or 200 mg.
Heart failure: PARADIGM-HF: PARADIGM-HF, the pivotal phase 3 study, was a multinational, randomised, double-blind study of 8,442 patients comparing sacubitril/valsartan to enalapril, both given to adult patients with chronic heart failure, NYHA class II-IV and reduced ejection fraction (left ventricular ejection fraction [LVEF] ≤40%, amended later to ≤35%) in addition to other heart failure therapy. The primary endpoint was the composite of cardiovascular (CV) death or hospitalisation for heart failure (HF). Patients with SBP <100 mmHg, severe renal impairment (eGFR <30 ml/min/1.73 m
2) and severe hepatic impairment were excluded at screening and therefore not prospectively studied.
Prior to study participation, patients were well treated with standard of care therapy which included ACE inhibitors/ARBs (>99%), beta blockers (94%), mineralocorticoid antagonists (58%) and diuretics (82%). The median follow-up duration was 27 months and patients were treated for up to 4.3 years.
Patients were required to discontinue their existing ACE inhibitor or ARB therapy and enter a sequential single-blind run-in period during which they received treatment with enalapril 10 mg twice daily, followed by single-blind treatment with sacubitril/valsartan 100 mg twice daily, increasing to 200 mg twice daily (see Adverse Reactions for discontinuations during this period). They were then randomised to the double-blind period of the study, during which they received either sacubitril/valsartan 200 mg or enalapril 10 mg twice daily [sacubitril/valsartan (n=4,209); enalapril (n=4,233)].
The mean age of the population studied was 64 years of age and 19% were 75 years of age or older. At randomisation, 70% of patients were NYHA class II, 24% were class III and 0.7% were class IV. The mean LVEF was 29% and there were 963 (11.4%) patients with a baseline LVEF >35% and ≤40%.
In the sacubitril/valsartan group, 76% of patients remained on the target dose of 200 mg twice daily at the end of the study (mean daily dose of 375 mg). In the enalapril group, 75% of patients remained on the target dose of 10 mg twice daily at the end of the study (mean daily dose of 18.9 mg).
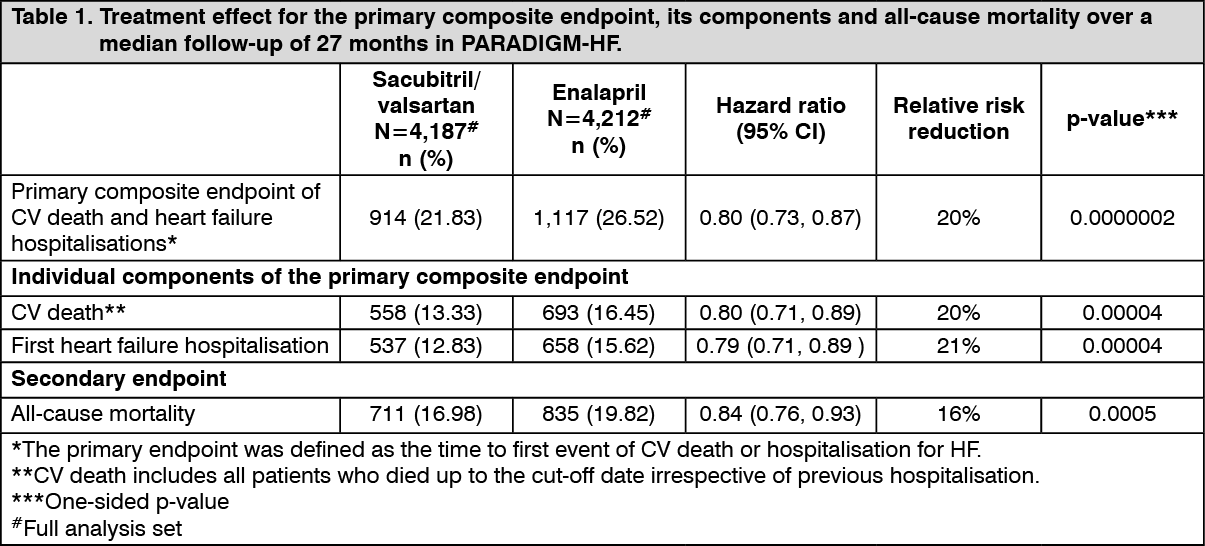
Sacubitril/valsartan was superior to enalapril, reducing the risk of cardiovascular death or heart failure hospitalisations to 21.8% compared to 26.5% for enalapril treated patients. The absolute risk reductions were 4.7% for the composite of the CV death or HF hospitalisation, 3.1% for CV death alone, and 2.8% for first HF hospitalisation alone. The relative risk reduction was 20% versus enalapril (see Table 1). This effect was observed early and was sustained throughout the duration of the study (see Figure 1). Both components contributed to the risk reduction. Sudden death accounted for 45% of cardiovascular deaths and was reduced by 20% in sacubitril/valsartan-treated patients compared to enalapril-treated patients (HR 0.80, p=0.0082). Pump failure accounted for 26% of cardiovascular deaths and was reduced by 21% in sacubitril/valsartan-treated patients compared to enalapril-treated patients (HR 0.79, p=0.0338).
This risk reduction was consistently observed across subgroups including: gender, age, race, geography, NYHA class (II/III), ejection fraction, renal function, history of diabetes or hypertension, prior heart failure therapy, and atrial fibrillation.
Sacubitril/valsartan improved survival with a significant reduction in all-cause mortality of 2.8% (sacubitril/valsartan, 17%, enalapril, 19.8%). The relative risk reduction was 16% compared with enalapril (see Table 1). (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
TITRATION: TITRATION was a 12-week safety and tolerability study in 538 patients with chronic heart failure (NYHA class II-IV) and systolic dysfunction (left ventricular ejection fraction ≤35%) naïve to ACE inhibitor or ARB therapy or on varying doses of ACE inhibitors or ARBs prior to study entry. Patients received a starting dose of sacubitril/valsartan of 50 mg twice daily and were up-titrated to 100 mg twice daily, then to the target dose of 200 mg twice daily, with either a 3-week or a 6-week regimen.
More patients who were naïve to previous ACE inhibitor or ARB therapy or on low-dose therapy (equivalent to <10 mg enalapril/day) were able to achieve and maintain sacubitril/valsartan 200 mg when up-titrated over 6 weeks (84.8%) versus 3 weeks (73.6%). Overall, 76% of patients achieved and maintained the target dose of sacubitril/valsartan 200 mg twice daily without any dose interruption or down-titration over 12 weeks.
PARAGON-HF: PARAGON-HF, was a multicenter, randomized, double-blind trial comparing Entresto and valsartan in 4,796 adult patients with symptomatic heart failure with left ventricular ejection fraction ≥45%, and structural heart disease [either left atrial enlargement (LAE) or left ventricular hypertrophy (LVH)]. Patients with a systolic blood pressure of <110 mmHg and patients with any prior echocardiographic LVEF < 40% at screening were excluded.
The primary objective of PARAGON-HF was to determine whether Entresto reduced the rate of the composite endpoint of total (first and recurrent) heart failure (HF) hospitalizations and cardiovascular (CV) death.
After discontinuing their existing ACE inhibitor or ARB therapy, patients entered sequential single-blind run-in periods during which they received valsartan 80 mg twice-daily, followed by Entresto 100 mg twice-daily. Patients on prior low doses of an ACEi or ARB began the run-in period receiving valsartan 40 mg twice-daily for 1-2 weeks. Patients who successfully completed the sequential run-in periods were randomized to receive either Entresto 200 mg (N = 2,419) twice-daily or valsartan 160 mg (N = 2,403) twice-daily. The median follow-up duration was 35 months and patients were treated for up to 4.7 years.
The population was 81% Caucasian, 13% Asian, and 2% Black; the mean age was 73 years and 52% were female. At randomization, 77% of patients were NYHA Class II, 19% were NYHA Class III, and 0.4% were NYHA Class IV. The median left ventricular ejection fraction was 57%. The underlying cause of heart failure was of ischemic etiology in 36% of patients. Furthermore, 96% had a history of hypertension, 23% had a history of myocardial infarction, 46% had an eGFR <60 mL/min/1.73 m
2, and 43% had diabetes mellitus. Most patients were taking beta-blockers (80%) and diuretics (95%).
PARAGON-HF demonstrated that Entresto had a numerical reduction in the rate of the composite endpoint of total (first and recurrent) HF hospitalizations and CV death, based on an analysis using a proportional rates model (rate ratio [RR] 0.87; 95% CI [0.75, 1.01], p = 0.06); see Table 2. The treatment effect was primarily driven by the reduction in total HF hospitalizations in patients randomized to Entresto (RR 0.85; 95% CI [0.72, 1.00]). (See Table 2.)
Click on icon to see table/diagram/image
Figure 2 shows the mean number of composite endpoint events of total HF hospitalizations and CV death over time. (See Figure 2.)
Click on icon to see table/diagram/image
A wide range of demographic characteristics, baseline disease characteristics, and baseline concomitant medications were examined for their influence on outcomes (Figure 3). (See Figure 3.)
Click on icon to see table/diagram/image
Note: The figure previously mentioned presents effects in various subgroups, all of which are baseline characteristics. The 95% confidence limits that are shown do not take into account the number of comparisons made, and may not reflect the effect of a particular factor after adjustment for all other factors.
In an analysis of the relationship between LVEF and outcome in PARADIGM-HF and PARAGON-HF, patients with LVEF below normal treated with Entresto experienced greater risk reduction (Figure 4). (See Figure 4.)
Click on icon to see table/diagram/image
Hypertension: The antihypertensive effect of Entresto was evaluated in two randomized, double-blind, active-controlled, 8-week studies evaluating the efficacy and safety of Entresto in comparison to olmesartan (CLCZ696A2315 and CLCZ696A1306) in more than 2,500 adult patients of which more than 1,700 patients received Entresto. Both studies demonstrated non-inferiority as well as superiority of the mean sitting systolic blood pressure (msSBP) lowering effect of both Entresto 200mg once daily (2.3 and 5.0 mmHg in each study, respectively) and Entresto 400mg once daily (3.5 and 7.0 mmHg) compared to olmesartan 20mg once daily. Consistent results were observed in mean diastolic BP.
Additionally, persistency of blood pressure lowering effect was demonstrated in a 52-week, safety, tolerability and efficacy, open-label, extension study (CLCZ696A2219E1) in which 341 patients were received Entresto as a monotherapy or in combination with amlodipine and hydrothiazide.
Pharmacokinetics: The valsartan contained within sacubitril/valsartan is more bioavailable than the valsartan in other marketed tablet formulations; 26 mg, 51 mg, and 103 mg of valsartan in sacubitril/valsartan is equivalent to 40 mg, 80 mg and 160 mg of valsartan in other marketed tablet formulations, respectively.
Absorption: Following oral administration, sacubitril/valsartan dissociates into valsartan and the prodrug sacubitril. Sacubitril is further metabolised to the active metabolite LBQ657. These reach peak plasma concentrations in 2 hours, 1 hour, and 2 hours, respectively. The oral absolute bioavailability of sacubitril and valsartan is estimated to be more than 60% and 23%, respectively.
Following twice daily dosing of sacubitril/valsartan, steady-state levels of sacubitril, LBQ657 and valsartan are reached in three days. At steady state, sacubitril and valsartan do not accumulate significantly, while LBQ657 accumulates 1.6-fold.
Following once daily dosing of Entresto, steady state levels of sacubitril, LBQ657 (sacubitrilat) and valsartan are achieved in 5 days with no accumulation in sacubitril and valsartan and 1.2-fold accumulation in sacubitrilat. Administration with food has no clinically significant impact on the systemic exposures of sacubitril, LBQ657 and valsartan. Sacubitril/valsartan can be administered with or without food.
Distribution: Sacubitril, LBQ657 and valsartan are highly bound to plasma proteins (94-97%). Based on the comparison of plasma and CSF exposures, LBQ657 crosses the blood brain barrier to a limited extent (0.28%). The average apparent volume of distribution of valsartan and sacubitril were 75 litres to 103 litres, respectively.
Biotransformation: Sacubitril is readily converted to LBQ657 by carboxylesterases 1b and 1c; LBQ657 is not further metabolised to a significant extent. Valsartan is minimally metabolised, as only about 20% of the dose is recovered as metabolites. A hydroxyl metabolite of valsartan has been identified in plasma at low concentrations (<10%).
Since CYP450-enzyme-mediated metabolism of sacubitril and valsartan is minimal, co-administration with medicinal products that impact CYP450 enzymes is not expected to impact the pharmacokinetics.
In vitro metabolism studies indicate that potential for CYP450-based drug interactions is low since there is limited metabolism of sacubitril/valsartan via CYP450 enzymes. Sacubitril/valsartan does not induce or inhibit CYP450 enzymes.
Elimination: Following oral administration, 52-68% of sacubitril (primarily as LBQ657) and ~13% of valsartan and its metabolites are excreted in urine; 37-48% of sacubitril (primarily as LBQ657) and 86% of valsartan and its metabolites are excreted in faeces.
Sacubitril, LBQ657 and valsartan are eliminated from plasma with a mean elimination half-life (T
½) of approximately 1.43 hours, 11.48 hours, and 9.90 hours, respectively.
Linearity/non-linearity: The pharmacokinetics of sacubitril, LBQ657 and valsartan were approximately linear over a sacubitril/valsartan dose range of 24 mg sacubitril/26 mg valsartan to 97 mg sacubitril/103 mg valsartan.
Special populations: Elderly patients: LBQ657 and valsartan exposure are increased in subjects over 65 years of age by 42% and 30%, respectively, compared to younger subjects.
Impaired renal function: A correlation was observed between renal function and systemic exposure to LBQ657 in patients with mild to severe renal impairment. The exposure of LBQ657 in patients with moderate (30 ml/min/1.73 m
2 ≤ eGFR <60 ml/min/1.73 m
2) and severe renal impairment (15 ml/min/1.73 m
2 ≤ eGFR <30 ml/min/1.73 m
2) was 1.4-fold and 2.2-fold higher compared to patients with mild renal impairment (60 ml/min/1.73 m
2 ≤ eGFR <90 ml/min/1.73 m
2), the largest group of patients enrolled in PARADIGM-HF). The exposure of valsartan was similar in patients with moderate and severe renal impairment compared to patients with mild renal impairment.
Safety and efficacy of Entresto in patients with essential hypertension and with severe renal impairment have not been established.
No studies have been performed in patients undergoing dialysis. However, LBQ657 and valsartan are highly bound to plasma protein and therefore unlikely to be effectively removed by dialysis.
Impaired hepatic function: In patients with mild to moderate hepatic impairment, the exposures of sacubitril increased by 1.5- and 3.4-fold, LBQ657 increased by 1.5- and 1.9-fold, and valsartan increased by 1.2-fold and 2.1-fold, respectively, compared to matching healthy subjects. However, in patients with mild to moderate hepatic impairment, the exposures of free concentrations of LBQ657 increased by 1.47- and 3.08-fold, respectively, and the exposures of free concentrations of valsartan increased by 1.09-fold and 2.20-fold, respectively, compared to matching healthy subjects.
In patients with moderate hepatic impairment (Child-Pugh B classification), a starting dose of 50 mg twice daily is recommended in patients with heart failure and 100 mg once daily in hypertensive patients.
Sacubitril/valsartan has not been studied in patients with severe hepatic impairment, biliary cirrhosis or cholestasis (see Contraindications and Precautions).
Effect of gender: The pharmacokinetics of sacubitril/valsartan (sacubitril, LBQ657 and valsartan) are similar between male and female subjects.
Race/Ethnicity: The pharmacokinetics of Entresto (sacubitril, sacubitrilat and valsartan) are comparable across different race and ethnic groups (Caucasians, Blacks, Asians, Japanese and others).
Toxicology: Preclinical safety data: Non-clinical data (including studies with sacubitril and valsartan components and/or sacubitril/valsartan) reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential and fertility.
Fertility, reproduction and development: Sacubitril/valsartan treatment during organogenesis resulted in increased embryofoetal lethality in rats at doses ≥49 mg sacubitril/51 mg valsartan/kg/day (≤0.72-fold the maximum recommended human dose [MRHD] on the basis of AUC) and rabbits at doses ≥4.9 mg sacubitril/5.1 mg valsartan/kg/day (2-fold and 0.03-fold the MRHD on the basis of valsartan and LBQ657 AUC, respectively). It is teratogenic based on a low incidence of foetal hydrocephaly, associated with maternally toxic doses, which was observed in rabbits at a sacubitril/valsartan dose of ≥4.9 mg sacubitril/5.1 mg valsartan/kg/day. Cardiovascular abnormalities (mainly cardiomegaly) were observed in rabbit foetuses at a maternally non-toxic dose (1.46 mg sacubitril/1.54 mg valsartan/kg/day). A slight increase in two foetal skeletal variations (misshapen sternebra, sternebra bipartite ossification) was observed in rabbits at a sacubitril/valsartan dose of 4.9 mg sacubitril/5.1 mg valsartan/kg/day. The adverse embryofoetal effects of sacubitril/valsartan are attributed to the angiotensin receptor antagonist activity (see Use in Pregnancy & Lactation).
Sacubitril treatment during organogenesis resulted in embryo-foetal lethality and embryo-foetal toxicity (decreased foetal body weights and skeletal malformations) in rabbits at doses associated with maternal toxicity (500 mg/kg/day; 5.7-fold the MRHD on the basis of LBQ657 AUC). A slight generalised delay in ossification was observed at doses of >50 mg/kg/day. This finding is not considered adverse. No evidence of embryo-foetal toxicity or teratogenicity was observed in rats treated with sacubitril. The embryo-foetal no-observed adverse effect level (NOAEL) for sacubitril was at least 750 mg/kg/day in rats and 200 mg/kg/day in rabbits (2.2-fold the MRHD on the basis of LBQ657 AUC).
Pre- and postnatal development studies in rats conducted with sacubitril at high doses up to 750 mg/kg/day (2.2-fold the MRHD on the basis of AUC) and valsartan at doses up to 600 mg/kg/day (0.86-fold the MRHD on the basis of AUC) indicate that treatment with sacubitril/valsartan during organogenesis, gestation and lactation may affect pup development and survival.
Other preclinical findings: Sacubitril/valsartan: The effects of sacubitril/valsartan on amyloid-β concentrations in CSF and brain tissue were assessed in young (2-4 years old) cynomolgus monkeys treated with sacubitril/valsartan (24 mg sacubitril/26 mg valsartan/kg/day) for two weeks. In this study CSF Aβ clearance in cynomolgus monkeys was reduced, increasing CSF Aβ1-40, 1-42 and 1-38 levels; there was no corresponding increase in Aβ levels in the brain. Increases in CSF Aβ1-40 and 1-42 were not observed in a two-week healthy volunteer study in humans (see Pharmacodynamics as previously mentioned). Additionally, in a toxicology study in cynomolgus monkeys treated with sacubitril/valsartan at 146 mg sacubitril/154 mg valsartan/kg/day for 39 weeks, there was no evidence for the presence of amyloid plaques in the brain. Amyloid content was not, however, measured quantitatively in this study.
Sacubitril: In juvenile rats treated with sacubitril (postnatal days 7 to 70), there was a reduction in age-related bone mass development and bone elongation. A study in adult rats showed only a minimal transient inhibitory effect on bone mineral density but not on any other parameters relevant for bone growth, suggesting no relevant effect of sacubitril on bone in adult patient populations under normal conditions. However, a mild transient interference of sacubitril with the early phase of fracture healing in adults cannot be excluded.
Valsartan: In juvenile rats treated with valsartan (postnatal days 7 to 70), doses as low as 1 mg/kg/day produced persistent irreversible kidney changes consisting of tubular nephropathy (sometimes accompanied by tubular epithelial necrosis) and pelvic dilatation. These kidney changes represent an expected exaggerated pharmacological effect of angiotensin converting enzyme inhibitors and angiotensin II type 1 blockers; such effects are observed if rats are treated during the first 13 days of life. This period coincides with 36 weeks of gestation in humans, which could occasionally extend up to 44 weeks after conception in humans.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image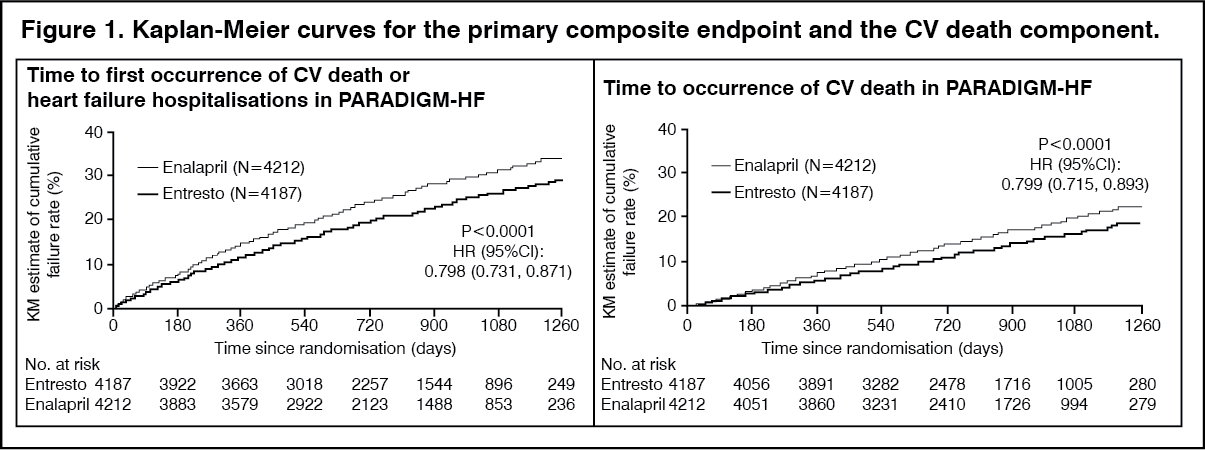 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image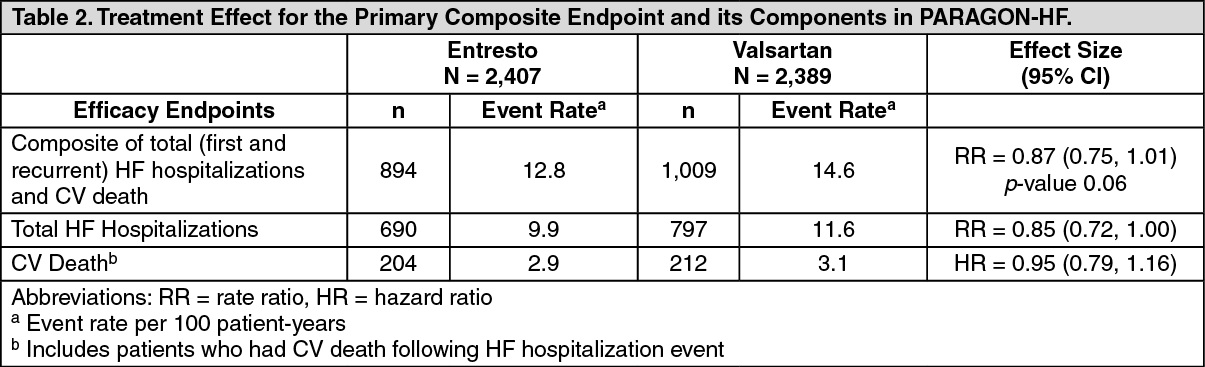 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image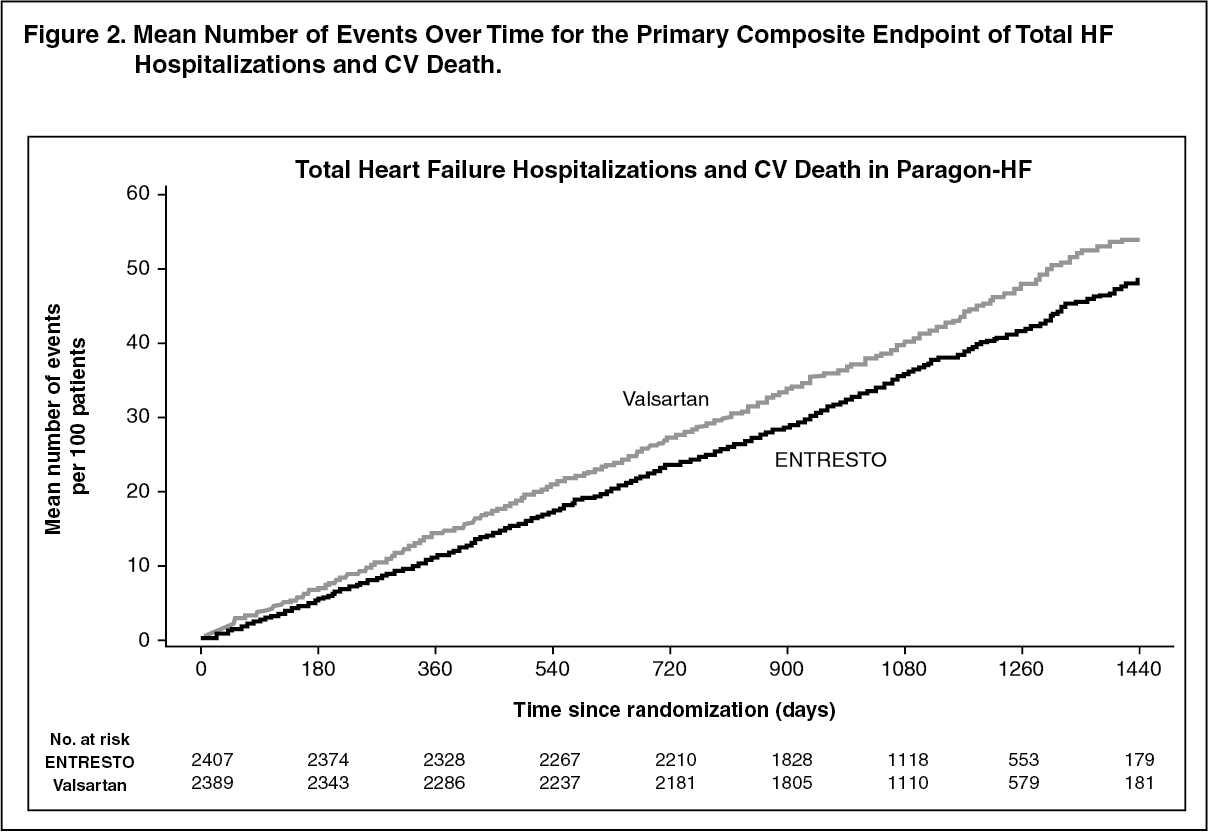 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image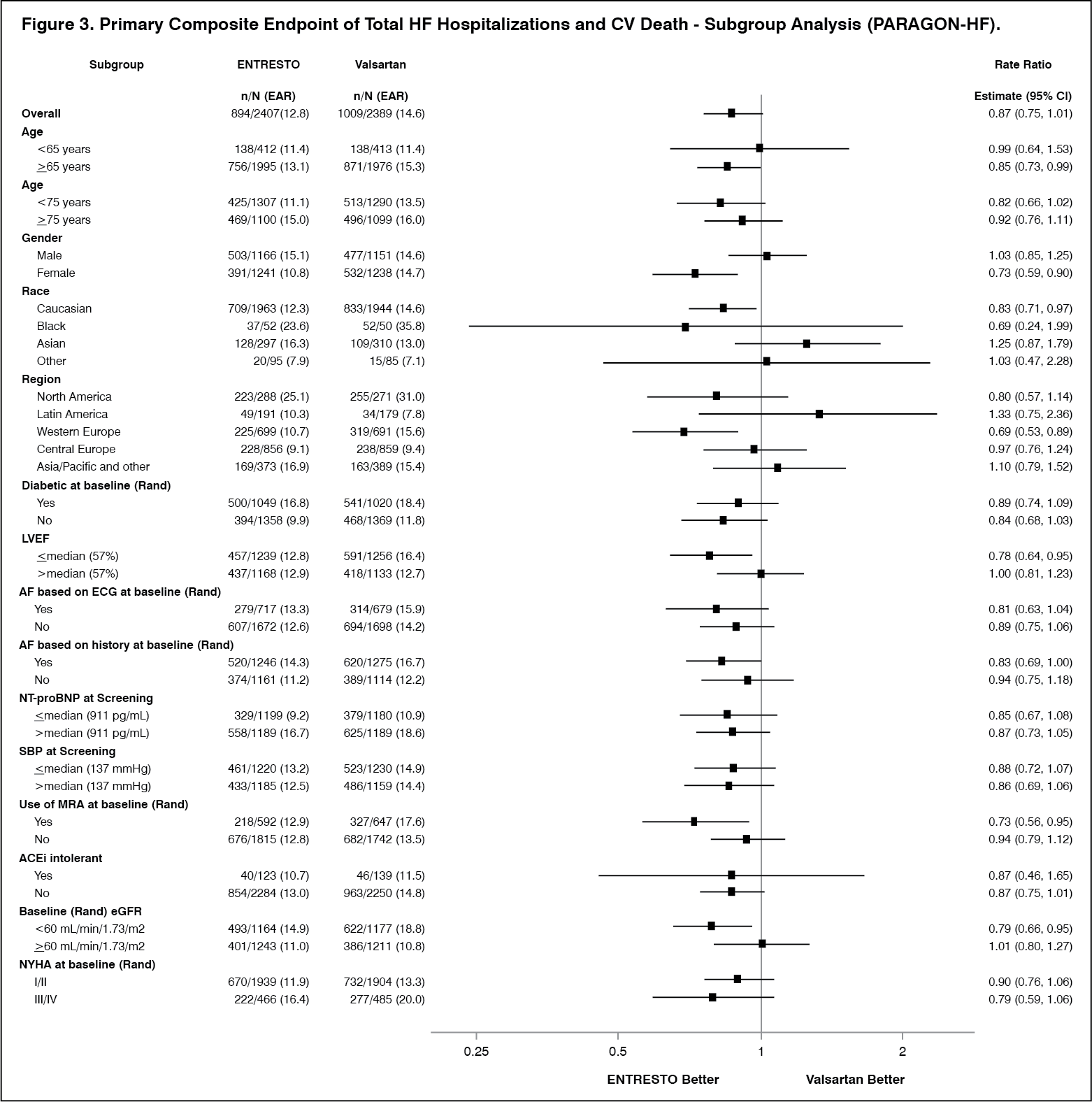 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image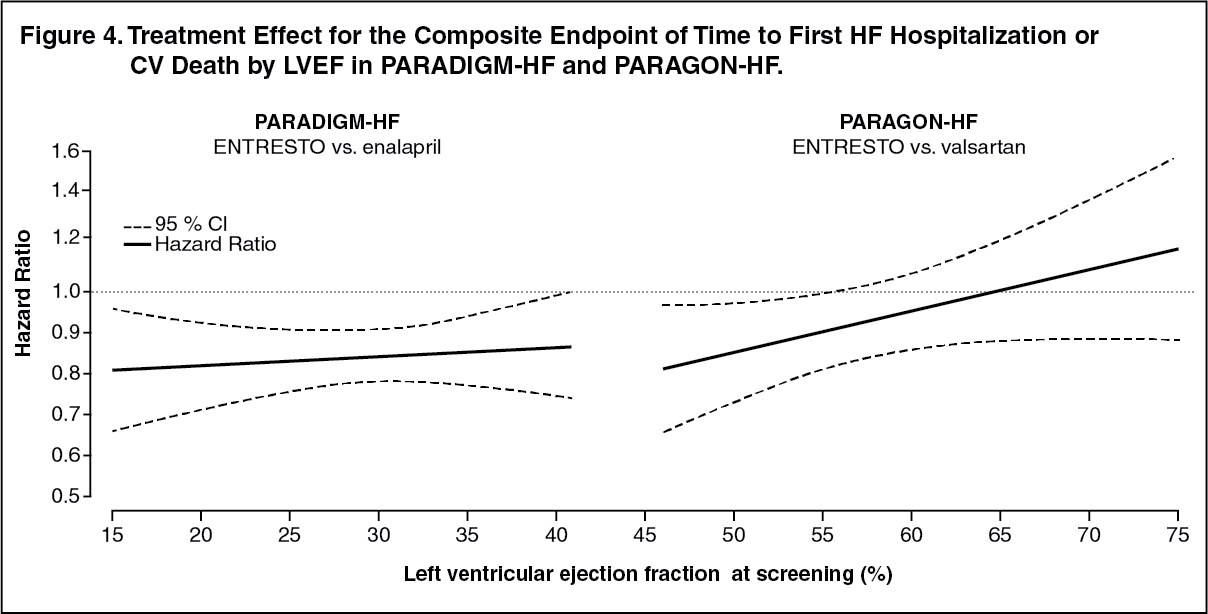 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image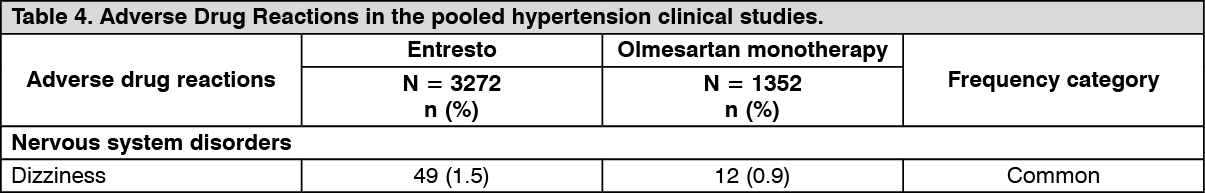 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out
