


 Sign Out
Sign Out


Pimecrolimus also prevents the release of cytokines and pro-inflammatory mediators from mast cells in vitro after stimulation by antigen/IgE. Pimecrolimus does not affect the growth of keratinocyte, fibroblast or endothelial cell lines and, in contrast to corticosteroids, does not impair the differentiation, maturation, functions and viability of murine Langerhans cells and human monocytes-derived dendritic cells, thus, underlining its cell-selective mode of action.
In studies using various topical formulations, including the pimecrolimus cream and tacrolimus ointment, pimecrolimus penetrates similarly into, but permeates less through skin in vitro than corticosteroids or tacrolimus, suggesting a lower systemic exposure to pimecrolimus after topical application as compared to tacrolimus and corticosteroids.
Pimecrolimus exhibits high anti-inflammatory activity in animal models of skin inflammation after topical and systemic application. Pimecrolimus is as effective as the high potency corticosteroids clobetasol-17-propionate and fluticasone after topical application in the pig model of allergic contact dermatitis (ACD). Topical pimecrolimus also inhibits the inflammatory response to irritants, as shown in murine models of irritant contact dermatitis.
Furthermore, topical and oral pimecrolimus effectively reduces skin inflammation and pruritus and normalises histopathological changes in hypomagnesemic hairless rats, a model that mimics acute aspects of atopic dermatitis. Oral pimecrolimus is superior to ciclosporin A by a factor of 4 and superior to tacrolimus by a factor of more than 2 in inhibiting skin inflammation in ACD of rats.
Topical pimecrolimus does not cause skin atrophy in pigs, unlike clobetasol-17-propionate. Furthermore, pimecrolimus does not cause blanching and changes in skin texture in pigs, unlike clobetasol-17-propionate and fluticasone.
Topical pimecrolimus does not affect epidermal Langerhans' cells in mice. In contrast, treatment with standard topical corticosteroids, including hydrocortisone, resulted in a reduction in Langerhans cells by 96 to 100%. A recent analysis of skin biopsies of atopic dermatitis patients has confirmed that treatment with the corticosteroid betamethasone 0.1 %, but not Elidel 1% cream, for 3 weeks results in depletion of Langerhans cells, while both drugs significantly reduce T cells. Thus, results from these as well as in vitro studies indicate that topically applied pimecrolimus is unlikely to interfere with the function of Langerhans/dendritic cells to differentiate naive T cells into effector T cells, which is key for the developing immune system and maintenance of specific immunocompetence.
In contrast to its efficacy in skin inflammation models, the potential of pimecrolimus for affecting systemic immune responses is lower than that of tacrolimus and ciclosporin A, as shown in models of systemic immunosuppression and based on dose comparison. In the rat, after subcutaneous administration, the potency of pimecrolimus in inhibiting the formation of antibodies is 48-fold lower than with tacrolimus. Subcutaneous injections of ciclosporin A and tacrolimus suppress the localized graft-versus-host reaction in rats 8-fold and 66-fold more potently than pimecrolimus. In contrast to ciclosporin A and tacrolimus, oral treatment of mice with pimecrolimus neither impairs the primary immune response nor decreases lymph node weight and cellularity in ACD.
The data show that topical pimecrolimus/Elidel has a high and selective anti-inflammatory activity in the skin and minimal percutaneous resorption. It differs from corticosteroids by its selective action on T cells and mast cells, by lack of impairment of Langerhans' cells/dendritic cells, by lack of induction of skin atrophy and by less permeation through skin.
It differs from tacrolimus by less permeation through skin and by a lower potential for affecting systemic immune responses.
In animal safety pharmacology studies, single oral doses of pimecrolimus had no effect on basal lung and cardiovascular functions. CNS and endocrine parameters (e.g. GH, prolactin, LH, testosterone, corticosterone) were also unaffected. Based on its mechanism of action as a selective inhibitor of the production and release of pro-inflammatory cytokines and mediators in T cells and mast cells, pimecrolimus is not expected to have any effect on the HPA axis.
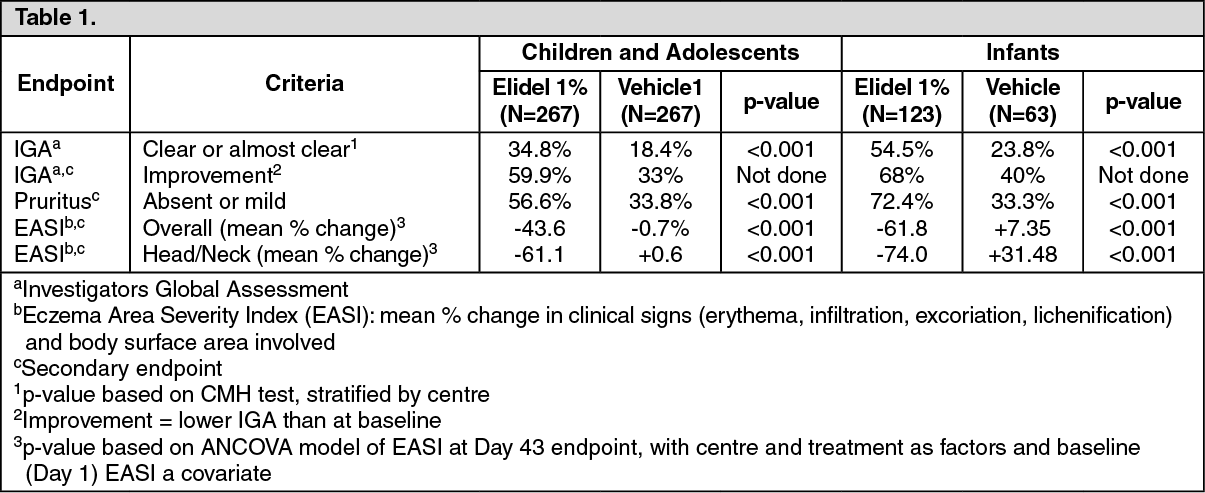
Clinical data: Short-term (acute) treatment in paediatric patients: Children and adolescents: Two 6-week, vehicle-controlled trials were conducted including a total of 403 paediatric patients aged 2 to 17 years. Patients were treated twice daily with Elidel 1% cream. The data of both studies were pooled.
Infants: A similar 6-week study was conducted in 186 patients aged 3 to 23 months.
In these three 6-week studies, the efficacy results at endpoint were as follows:
A significant improvement in pruritus was observed within the first week of treatment in 44% of children and adolescents and in 70% of infants.
Long-term treatment in paediatric patient: In two double-blind studies of long-term management of atopic dermatitis in 713 children and adolescents (2 to 17 years) and 251 infants (3 to 23 months), Elidel 1% cream was evaluated as first line foundation therapy.
In addition to emollients, the Elidel group received Elidel 1% cream used at first signs of itching and redness to prevent progression to flares of atopic dermatitis. Only in case of flare not controlled by Elidel 1% cream, treatment with medium potency topical corticosteroids was initiated.
The control group received a standard treatment consisting of emollient plus medium potency topical corticosteroids to treat flares. Elidel vehicle was used instead of Elidel 1% cream in order to maintain the studies blind.
Both studies showed a reduction in the incidence of flares (p <0.001) in favour of Elidel 1% cream first-line treatment; Elidel 1% cream first-line treatment showed better efficacy in all secondary assessments (Eczema Area Severity Index, IGA, subject assessment); pruritus was controlled within a week with Elidel 1% cream. Significantly more patients on Elidel 1% cream completed 6 months (children (61% Elidel 1% cream vs 34% control); infants (70% Elidel vs 33% control) and 12 months (children 51% Elidel vs 28% control) with no flare.
Significantly more patients treated with Elidel 1% cream did not use corticosteroids in the first 6 months (children: 65% Elidel vs 37% control; infants: 70% Elidel vs 39% control) or 12 months (children: 57% Elidel 1% cream vs 32% control). The efficacy of Elidel 1% cream was maintained over time with the ability to prevent disease progression to severe flares.
Special studies: Tolerability studies demonstrated that Elidel 1% cream is devoid of any irritation, contact sensitising, phototoxic or photosensitising potential.
The atrophogenic potential of Elidel 1% cream in humans was tested in comparison to medium and highly potent topical steroids (betamethasone-17-valerate 0.1% cream, triamcinolone acetonide 0.1% cream) and vehicle in sixteen healthy volunteers treated for 4 weeks. Both topical corticosteroids, induced a significant reduction in skin thickness measured by echography, as compared to Elidel 1% cream and vehicle, which did not induce a reduction of skin thickness. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Data in animals: Pimecrolimus is lipophilic. When applied topically its permeation through skin is very low. In minipigs, the total drug-related material systemically absorbed following a single 22 h application of Elidel 1% cream under semi-occlusion was at most 1% of the dose; the bioavailability of unchanged pimecrolimus was estimated to be about 0.03%. The amount of radiolabeled drug-related material in the skin at the application site remained essentially constant in the time interval 0 to 10 days after a 22-hour application; at 5 days post-dose, it represented almost exclusively unchanged pimecrolimus. The major fraction of the absorbed topical dose was completely metabolised and excreted slowly via the bile into the faeces.
Data in humans: Absorption in adults: Systemic exposure to pimecrolimus was investigated in 12 adult patients treated with Elidel 1% cream twice daily for 3 weeks. These patients had atopic dermatitis (eczema) lesions affecting 15 to 59% of their body surface area (BSA). 77.5% of pimecrolimus blood concentrations were below 0.5 ng/mL, the assay limit of quantitation (LoQ), and 99.8% of the total samples were below 1 ng/mL. The highest blood concentration of pimecrolimus measured in one patient was 1.4 ng/mL.
In 40 adult patients treated for up to 1 year with Elidel, having 14 to 62% of their BSA affected at baseline, 98% of pimecrolimus blood concentrations of pimecrolimus were consistently low, mostly below the LoQ. A maximum blood concentration of 0.8 ng/mL was measured in only 2 patients in week 6 of treatment. There was no increase in blood concentration over time in any patient during the 12 months of treatment. In 13 adult patients with hand dermatitis treated with Elidel twice daily for 3 weeks (palmar and dorsal surfaces of hands treated, overnight occlusion), the maximum blood concentration of pimecrolimus measured was 0.91 ng/mL.
Given the high proportion of pimecrolimus blood levels below the LoQ after topical application, the AUC could only be calculated from a few individuals. In 8 adult AD patients presenting with at least three quantifiable blood levels per visit day, the AUC(0 to 12h) values ranged from 2.5 to 11.4 ng x h/mL.
Absorption in children: Systemic exposure to pimecrolimus was investigated in 58 paediatric patients aged 3 months to 14 years, who had atopic dermatitis (eczema) lesions involving 10 to 92% of the total body surface area. These children were treated with Elidel 1% cream twice daily for 3 weeks and five out of them were treated for up to 1 year on a "as needed" basis.
The blood concentrations measured in these paediatric patients were consistently low regardless of the extent of lesions treated or duration of therapy. They were in a range similar to that measured in adult patients treated under the same dosing regimen. 60% of pimecrolimus blood concentrations were below 0.5 ng/mL (LoQ) and 97% of all samples were below 2 ng/mL. The highest blood concentrations measured in 2 paediatric patients aged 8 months to 14 years of age were 2.0 ng/mL.
In the youngest patients (aged 3 to 23 months), the highest blood concentration measured in one patient was 2.6 ng/mL. In the 5 children treated for 1 year, blood concentrations were consistently low, and the maximum blood concentration measured was 1.94 ng/mL (1 patient). In these five patients, there was no increase in blood concentration over time in any patient during the 12 months of treatment.
In 8 paediatric patients aged 2-14 years presenting at least three measurable blood concentrations per visit day, AUC(0 to 12h) ranged from 5.4 to 18.8 ng x h/mL. AUC ranges observed in patients with <40% BSA affected at baseline were comparable to those in patients with ≥40% BSA.
Comparison to oral PK Data: In psoriatic patients treated with oral pimecrolimus doses ranging from 5 mg once daily to 30 mg twice daily for 4 weeks, the drug was well tolerated at all doses including the highest dose. No significant adverse events were reported and no significant change was observed in physical examination, vital signs, and laboratory (including renal) safety parameters. The highest dose was associated with an AUC(0 to 12h) of 294.9 ng x h/mL. This exposure is approximately 26 and 16 times higher, respectively, than the highest systemic exposure observed in adult and paediatric atopic dermatitis (eczema) patients treated topically with Elidel twice daily for 3 weeks (AUC(0 to 12h) of 11.4 ng x h/mL and 18.8 ng x h/mL, respectively).
Distribution: Consistent with its skin selectivity, after topical application, pimecrolimus blood levels are very low. Therefore, pimecrolimus metabolism could not be determined after topical administration.
In vitro plasma protein binding studies have shown that 99.6% of pimecrolimus in plasma is bound to proteins. The major fraction of pimecrolimus in plasma is bound to different lipoproteins.
Metabolism: After single oral administration of radiolabeled pimecrolimus in healthy subjects, unchanged pimecrolimus was the major drug-related component in blood and there were numerous minor metabolites of moderate polarity that appeared to be products of O-demethylations and oxygenation.
No drug metabolism was observed in human skin in vitro.
Elimination: Drug-related radioactivity was excreted principally via the faeces (78.4%) and only a small fraction (2.5%) was recovered in urine. Total mean recovery of radioactivity was 80.9%. Parent compound was not detected in urine and less than 1% of radioactivity in faeces was accounted for by unchanged pimecrolimus.
Toxicology: Preclinical Safety Data: Toxicology studies after dermal application: A variety of preclinical safety studies were conducted with the pimecrolimus cream formulations in several animal species. There was no evidence of irritation, (photo) sensitisation, or local or systemic toxicity.
In a 2-year dermal carcinogenicity study in rats using Elidel 1% cream, no cutaneous or systemic carcinogenic effects were observed up to the highest practicable dose of 10 mg/kg/day or 110 mg/m2/day, represented by a mean AUC(0 to 24h) of 125 ng x h/mL (equivalent to 3.3 times the maximum exposure observed in paediatric patients in clinical trials). In a mouse dermal carcinogenicity study using pimecrolimus in an ethanolic solution, no increase in incidence of neoplasms was observed in the skin or other organs up to the highest dose of 4 mg/kg/day or 12 mg/m2/day, corresponding to a mean AUC(0 to 24h) value of 1,040 ng x h/mL (equivalent to 27 times the maximum exposure observed in paediatric patients in clinical trials).
In a dermal photocarcinogenicity study in hairless mice using Elidel 1% cream, no photocarcinogenic effect versus vehicle treated animals was noted up to the highest dose of 10 mg/kg/day or 30 mg/m2/day, corresponding to a mean AUC(0-24h) value of 2,100 ng x h/mL (equivalent to 55 times the maximum exposure observed in pediatric patients in clinical trials).
In dermal reproduction studies, no maternal or fetal toxicity was observed up to the highest practicable doses tested, 10 mg/kg/day or 110 mg/m2/day in rats and 10 mg/kg/day or 36 mg/m2/day in rabbits. In rabbits, the corresponding mean AUC(0 to 24h) was 24.8 ng x h/mL. AUC could not be calculated in rats.
Toxicology studies after oral administration: Adverse reactions not observed in clinical studies but seen in animals at exposures considered sufficiently in excess of the maximum human exposure, indicating little relevance to clinical use, were as follows: reproduction studies in rats receiving oral doses up to 45 mg/kg/day or 490 mg/m2/day, corresponding to an extrapolated mean AUC (0 to 24h) of 1448 ng x h/mL (equivalent to at least 63 times the maximum exposure observed in adult patients), revealed slight maternal toxicity, oestrus cycle disturbances, post-implantation loss and reduction in litter size.
An oral fertility and embryo-foetal developmental study in rats revealed estrus cycle disturbances, post-implantation loss and reduction in litter size at the 45 mg/kg/day dose (38 times the Maximum Recommended Human Dose (MRHD) based on AUC comparisons). No effect on fertility in female rats was noted at 10 mg/kg/day (12 x MRHD based on AUC comparisons). No effect on fertility in male rats was noted at 45 mg/kg/day (23 x MRHD based on AUC comparisons), which was the highest dose tested in this study.
A second oral fertility and embryo-foetal developmental study in rats revealed reduced testicular and epididymal weights, reduced testicular sperm counts and motile sperm for males and estrus cycle disturbances, decreased corpora lutea, decreased implantations and viable fetuses for females at 45 mg/kg/day dose (123 x MRHD for males and 192 x MRHD for females based on AUC comparisons). No effect on fertility in female rats was noted at 10 mg/kg/day (5 x MRHD based on AUC comparisons). No effect on fertility in male rats was noted at 2 mg/ kg/day (0.7 x MRHD based on AUC comparisons). In an oral reproduction study in rabbits, maternal toxicity, but no embryotoxicity or teratogenicity was observed at the highest dose of 20 mg/kg/day or 72 mg/m2/day corresponding to an extrapolated mean AUC(0 to 24h) value of 147 ng x h/mL (equivalent to at least 6 times the maximum exposure observed in adult patients).
In a mouse oral carcinogenicity study, a 13% higher incidence of lymphomas versus controls associated with signs of immunosuppression was observed at 45 mg/kg/day or 135 mg/m2/day, corresponding to a mean AUC(0 to 24h) value of 9,821 ng x h/mL (equivalent to at least 258 times the maximum exposure observed in paediatric patients in clinical trials). A dose of 15 mg/kg/day or 45 mg/m2/day, corresponding to a mean AUC(0 to 24h) value of 5,059 ng x h/mL, produced no lymphomas or discernible effects on the immune system (equivalent to 133 times the maximum exposure observed in paediatric patients in clinical trials). In an oral rat carcinogenicity study, no carcinogenic potential was observed up to a dose of 10 mg/kg/day or 110 mg/m2/day, exceeding the maximum tolerated dose, represented by a mean AUC(0 to 24h) value of 1550 ng x h/mL (equivalent to 41 times the maximum exposure observed in pediatric patients in clinical trials).
In a 39-week monkey oral toxicity study, a dose-related immunosuppressive-related lymphoproliferative disorder (IRLD) associated with lymphocryptovirus (LCV) and other opportunistic infections were observed, beginning at 15 mg/kg/day, corresponding to a mean AUC(0-24) value of 1,193 ng x h/ mL (31 times the maximum exposure observed in pediatric patients in clinical trials). At 45 mg/kg/day, corresponding to a mean AUC(0-24) value of 3,945 ng x h/mL (104 times the maximum exposure observed in pediatric patients in clinical trials) IRLD was accompanied by mortality/moribundity, food consumption and body weight reductions, and pathological changes secondary to compound-related immunosuppression. Recovery and/or at least partial reversibility of the effects were noted upon cessation of dosage.
A battery of in vitro and in vivo genotoxicity texts, including the Ames assay, mouse lymphoma L5178Y assay, chromosome aberration test in V79 Chinese hamster cells, and mouse micronucleus test revealed no evidence for a mutagenic or clastogenic potential of the drug.