Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another product and may not reflect the rates observed in practice.
The following serious adverse reactions are described as follows and elsewhere in the labeling: Hypersensitivity Reactions Including Anaphylaxis [see Precautions].
In clinical trials, the most common adverse reactions (>10%) following ELAPRASE treatment were hypersensitivity reactions, and included rash, urticaria, pruritus, flushing, pyrexia, and headache. Most hypersensitivity reactions requiring intervention were ameliorated with slowing of the infusion rate, temporarily stopping the infusion, with or without administering additional treatments including antihistamines, corticosteroids or both prior to or during infusions.
In clinical trials, the most frequent serious adverse reactions following ELAPRASE treatment were hypoxic episodes. Other notable serious adverse reactions that occurred in the ELAPRASE-treated patients but not in the placebo-treated patients included one case each of: cardiac arrhythmia, pulmonary embolism, cyanosis, respiratory failure, infection, and arthralgia.
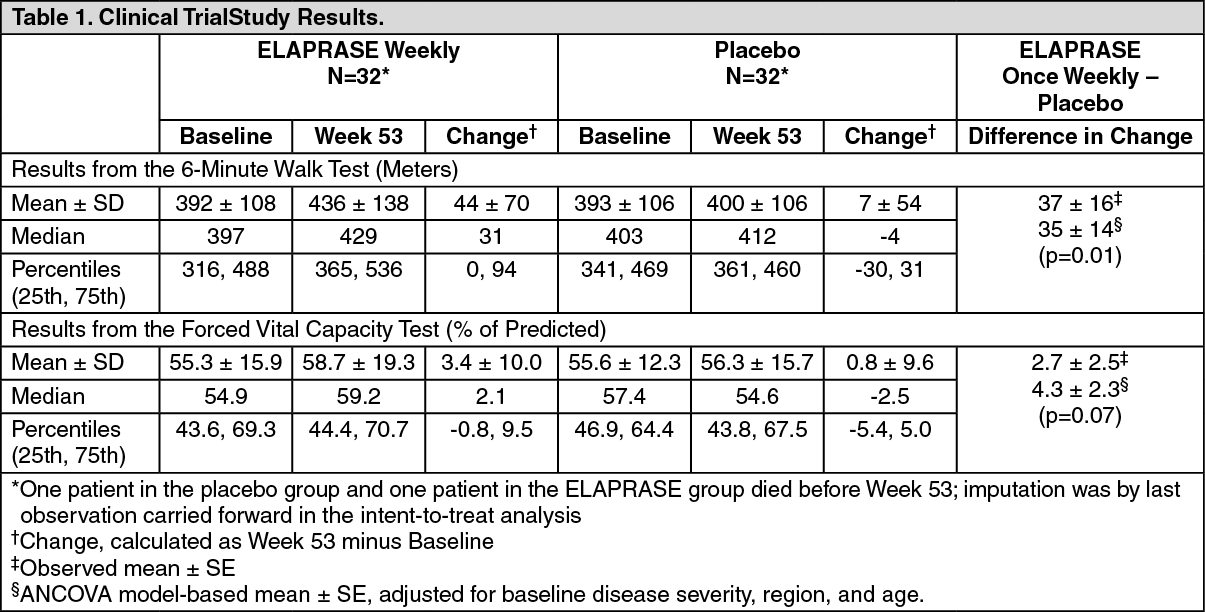
Clinical Trials in Patients 5 Years and Older: A 53-week, double-blind, placebo-controlled clinical trial of ELAPRASE was conducted in 96 male patients with Hunter syndrome, ages 5-31 years old. Of the 96 patients, 83% were White, non-Hispanic. Patients were randomized to three treatment groups, each with 32 patients: ELAPRASE 0.5 mg/kg once weekly, ELAPRASE 0.5 mg/kg every other week, or placebo. Hypersensitivity reactions were reported in 69% (22 of 32) of patients who received once-weekly treatment of ELAPRASE.
Table 4 summarizes the adverse reactions that occurred in at least 9% of patients (≥3 patients) in the ELAPRASE 0.5 mg/kg once weekly group and with a higher incidence than in the placebo group. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Additional adverse reactions that occurred in at least 9% of patients (≥3 patients) in the ELAPRASE 0.5 mg/kg every other week group and with a higher incidence than in the placebo group included: rash (19%), flushing (16%), fatigue (13%), tachycardia (9%), and chills (9%).
Extension Trial: An open-label extension trial was conducted in patients who completed the placebo-controlled trial. Ninety-four of the 96 patients who were enrolled in the placebo-controlled trial consented to participate in the extension trial. All 94 patients received ELAPRASE 0.5 mg/kg once weekly for 24 months. No new serious adverse reactions were reported. Approximately half (53%) of patients experienced hypersensitivity reactions during the 24-month extension trial. In addition to the adverse reactions listed in Table 4, common hypersensitivity reactions occurring in at least 5% of patients (≥ 5 patients) in the extension trial included: rash (23%), pyrexia (9%), flushing (7%), erythema (7%), nausea (5%), dizziness (5%), vomiting (5%), and hypotension (5%).
Clinical Trial in Patients 7 Years and Younger
: A 53-week, open-label, single-arm, safety trial of once weekly ELAPRASE 0.5 mg/kg treatment was conducted in patients with Hunter syndrome, ages 16 months to 4 years old (n=20) and ages 5 to 7.5 years old (n=8) at enrollment. Patients experienced similar adverse reactions as those observed in clinical trials in patients 5 years and older, with the most common adverse reactions following ELAPRASE treatment being hypersensitivity reactions (57%). A higher incidence of the following common hypersensitivity reactions were reported in this younger age group: pyrexia (36%), rash (32%) and vomiting (14%). The most common serious adverse reactions occurring in at least 10% of patients (≥ 3 patients) included: bronchopneumonia/pneumonia (18%), ear infection (11%), and pyrexia (11%).
Twenty-seven patients had results of genotype analysis: 15 patients had complete gene deletion, large gene rearrangement, nonsense, frameshift or splice site mutations and 12 patients had missense mutations.
Safety results demonstrated that patients with complete gene deletion, large gene rearrangement, nonsense, frameshift, or splice site mutations are more likely to experience hypersensitivity reactions and have serious adverse reactions following ELAPRASE administration, compared to patients with missense mutations. Table 5 summarizes these findings. (See Table 5.)
Click on icon to see table/diagram/image
Immunogenicity: Clinical Trials in Patients 5 Years and Older: As with all therapeutic proteins, there is potential for immunogenicity. In clinical trials in patients 5 years and older, 63 of the 64 patients treated with ELAPRASE 0.5 mg/kg once weekly or placebo for 53 weeks, followed by ELAPRASE 0.5 mg/kg once weekly in the extension trial, had immunogenicity data available for analysis. Of the 63 patients, 32 (51%) patients tested positive for anti-idursulfase IgG antibodies (Ab) at least one time (Table 5). Of the 32 Ab-positive patients, 23 (72%) tested positive for Ab at three or more different time points (persistent Ab). The incidence of hypersensitivity reactions was higher in patients who tested positive for Ab than those who tested negative.
Thirteen of 32 (41%) Ab-positive patients also tested positive for antibodies that neutralize idursulfase uptake into cells (uptake neutralizing antibodies, uptake NAb) or enzymatic activity (activity NAb) at least one time, and 8 (25%) of Ab-positive patients had persistent NAb. There was no clear relationship between the presence of either Ab or NAb and therapeutic response.
Clinical Trial in Patients 7 Years and Younger: In the clinical trial in patients 7 years and younger, 19 of 28 (68%) patients treated with ELAPRASE 0.5 mg/kg once weekly tested Ab-positive. Of the 19 Ab-positive patients, 16 (84%) tested positive for Ab at three or more different time points (persistent Ab). In addition, 15 of 19 (79%) Ab-positive patients tested positive for NAb, with 14 of 15 (93%) NAb-positive patients having persistent NAb.
All 15 patients with complete gene deletion, large gene rearrangement, nonsense, frameshift or splice site mutations tested positive for Ab (Table 5). Of these 15 patients, neutralizing antibodies were observed in 13 (87%) patients. The NAbs in these patients developed earlier (most reported to be positive at Week 9 rather than at Week 27, as reported in clinical trials in patients older than 5 years of age) and were associated with higher titers and greater in vitro neutralizing activity than in patients older than 5 years of age. The presence of Ab was associated with reduced systemic idursulfase exposure [see Pharmacology under Actions].
The immunogenicity data reflect the percentage of patients whose test results were positive for antibodies to idursulfase in specific assays, and are highly dependent on the sensitivity and specificity of these assays. The observed incidence of positive antibody in an assay may be influenced by several factors, including sample handling, timing of sample collection, concomitant medication, and underlying disease. For these reasons, comparison of the incidence of antibodies to idursulfase with the incidence of antibodies to other products may be misleading.
Postmarketing Experience: The following adverse reactions have been identified during post approval use of ELAPRASE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In post-marketing experience, late-emergent symptoms and signs of anaphylactic reactions have occurred up to 24 hours after initial treatment and recovery from an initial anaphylactic reaction. In addition, patients experienced repeated anaphylaxis over a two- to four-month period, up to several years after initiating ELAPRASE treatment [see Precautions].
A seven year-old male patient with Hunter syndrome, who received ELAPRASE at twice the recommended dosage (1 mg/kg weekly) for 1.5 years, experienced two anaphylactic events after 4.5 years of treatment. Treatment has been withdrawn [see Overdosage].
Serious adverse reactions that resulted in death included cardiorespiratory arrest, respiratory failure, respiratory distress, cardiac failure, and pneumonia.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image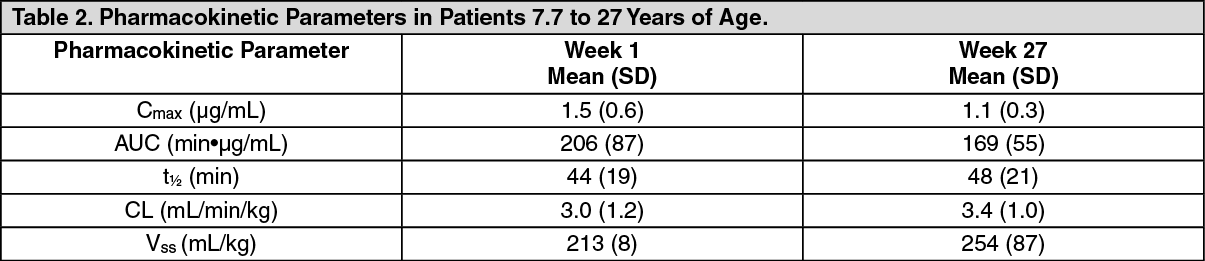 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image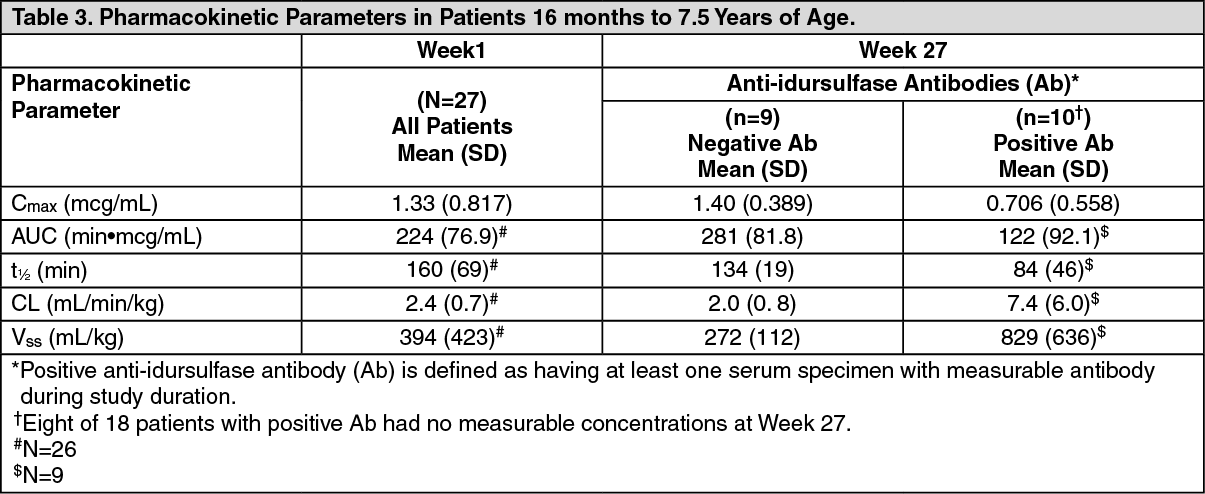 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image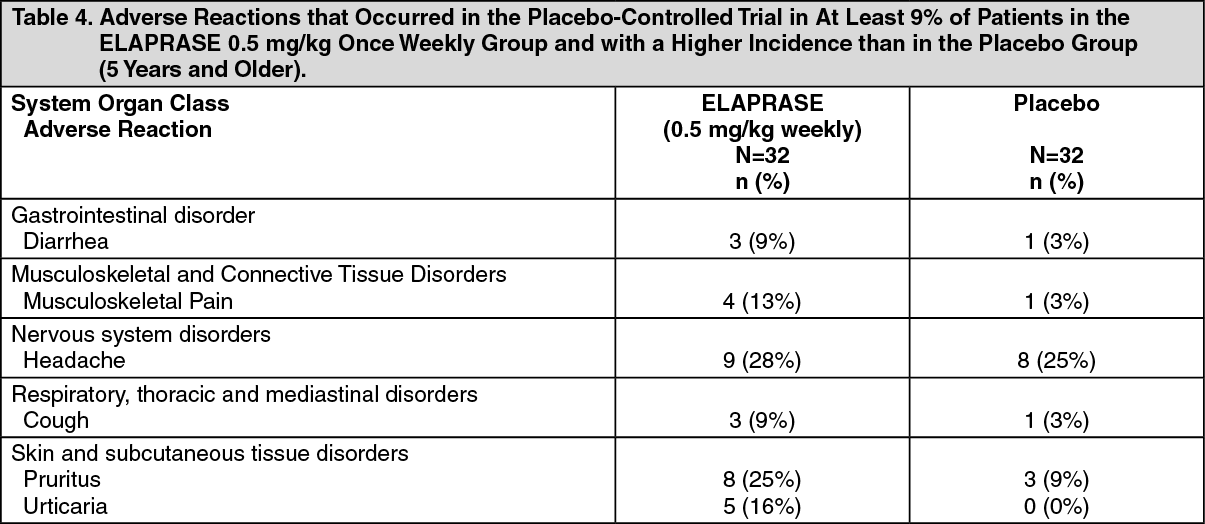 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image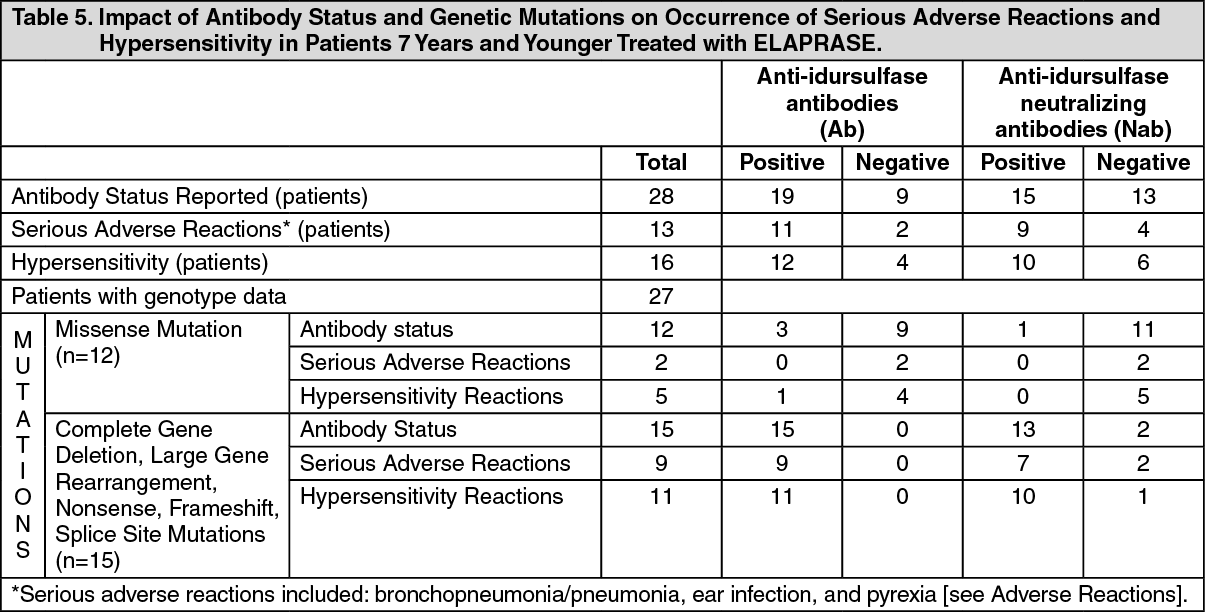 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
