


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Consistent with inhibition of IL-4 and IL-13 signaling, dupilumab treatment decreased certain biomarkers. In asthma subjects, fractional exhaled nitric oxide (FeNO) and circulating concentrations of eotaxin-3, total IgE, allergen specific IgE, TARC, and periostin were decreased relative to placebo. These reductions in biomarkers were comparable for the 300 mg Q2W and 200 mg Q2W regimens. These markers were near maximal suppression after 2 weeks of treatment, except for IgE which declined more slowly. These effects were sustained throughout treatment. The median percent reduction from baseline in total IgE concentrations with dupilumab treatments was 52% at Week 24 (DRI12544) and 70% at Week 52 (QUEST). For FeNO, the mean percent reduction from baseline at Week 2 was 35% and 24% in DRI12544 and QUEST respectively, and in the overall safety population, the mean FeNO level decreased to 20 ppb.
Mechanism of Action: Dupilumab is a human monoclonal IgG4 antibody that inhibits interleukin-4 (IL-4) and interleukin-13 (IL-13) signaling by specifically binding to the IL-4Rα subunit shared by the IL-4 and IL-13 receptor complexes. Dupilumab inhibits IL-4 signaling via the Type I receptor and both IL-4 and IL-13 signaling through the Type II receptor.
Inflammation is an important component in the pathogenesis of asthma and atopic dermatitis. Multiple cell types that express IL-4Rα (e.g., mast cells, eosinophils, macrophages, lymphocytes, epithelial cells, goblet cells) and inflammatory mediators (e.g., histamine, eicosanoids, leukotrienes, cytokines, chemokines) are involved in inflammation.
Blocking IL-4Rα with dupilumab inhibits IL-4 and IL-13 cytokine-induced inflammatory responses, including the release of proinflammatory cytokines, chemokines, nitric oxide and IgE; however, the mechanism of dupilumab action in asthma has not been definitively established.
Clinical Studies: Atopic Dermatitis: Adults with Atopic Dermatitis: Three randomized, double-blind, placebo-controlled trials (SOLO 1, SOLO 2, and CHRONOS; NCT02277743, NCT02277769, and NCT02260986 respectively) enrolled a total of 2119 subjects 18 years of age and older with moderate-to-severe atopic dermatitis (AD) not adequately controlled by topical medication(s). Disease severity was defined by an Investigator's Global Assessment (IGA) score ≥3 in the overall assessment of AD lesions on a severity scale of 0 to 4, an Eczema Area and Severity Index (EASI) score ≥16 on a scale of 0 to 72, and a minimum body surface area involvement of ≥10%. At baseline, 59% of subjects were male, 67% were white, 52% of subjects had a baseline IGA score of 3 (moderate AD), and 48% of subjects had a baseline IGA of 4 (severe AD). The baseline mean EASI score was 33 and the baseline weekly averaged peak pruritus Numeric Rating Scale (NRS) was 7 on a scale of 0-10.
In all three trials, subjects in the DUPIXENT group received subcutaneous injections of DUPIXENT 600 mg at Week 0, followed by 300 mg every other week (Q2W). In the monotherapy trials (SOLO 1 and SOLO 2), subjects received DUPIXENT or placebo for 16 weeks.
In the concomitant therapy trial (CHRONOS), subjects received DUPIXENT or placebo with concomitant topical corticosteroids (TCS) and as needed topical calcineurin inhibitors for problem areas only, such as the face, neck, intertriginous and genital areas for 52 weeks.
All three trials assessed the primary endpoint, the change from baseline to Week 16 in the proportion of subjects with an IGA 0 (clear) or 1 (almost clear) and at least a 2-point improvement. Other endpoints included the proportion of subjects with EASI-75 (improvement of at least 75% in EASI score from baseline) and reduction in itch as defined by at least a 4-point improvement in the peak pruritus NRS from baseline to Week 16.
Clinical Response at Week 16 (SOLO 1, SOLO 2, and CHRONOS): The results of the DUPIXENT monotherapy trials (SOLO 1 and SOLO 2) and the DUPIXENT with concomitant TCS trial (CHRONOS) are presented in Table 1. (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn CHRONOS, of the 421 subjects, 353 had been on study for 52 weeks at the time of data analysis. Of these 353 subjects, responders at Week 52 represent a mixture of subjects who maintained their efficacy from Week 16 (e.g., 53% of DUPIXENT IGA 0 or 1 responders at Week 16 remained responders at Week 52) and subjects who were non-responders at Week 16 who later responded to treatment (e.g., 24% of DUPIXENT IGA 0 or 1 non-responders at Week 16 became responders at Week 52). Results of supportive analyses of the 353 subjects in the DUPIXENT with concomitant TCS trial (CHRONOS) are presented in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment effects in subgroups (weight, age, gender, race, and prior treatment, including immunosuppressants) in SOLO 1, SOLO 2, and CHRONOS were generally consistent with the results in the overall study population.
In SOLO 1, SOLO 2, and CHRONOS, a third randomized treatment arm of DUPIXENT 300 mg QW did not demonstrate additional treatment benefit over DUPIXENT 300 mg Q2W.
Subjects in SOLO 1 and SOLO 2 who had an IGA 0 or 1 with a reduction of ≥2 points were re-randomized into SOLO CONTINUE (NCT02395133). SOLO CONTINUE evaluated multiple DUPIXENT monotherapy dose regimens for maintaining treatment response. The study included subjects randomized to continue with DUPIXENT 300 mg Q2W (62 subjects) or switch to placebo (31 subjects) for 36 weeks. IGA 0 or 1 responses at Week 36 were as follows: 33 (53%) in the Q2W group and 3 (10%) in the placebo group.
Pediatric Subjects (12 to 17 Years of Age) with Atopic Dermatitis: The efficacy and safety of DUPIXENT monotherapy in pediatric subjects 12 to 17 years of age was evaluated in a multicenter, randomized, double-blind, placebo-controlled trial (AD-1526; NCT03054428) in 251 pediatric subjects 12 to 17 years of age, with moderate-to-severe AD defined by an IGA score ≥3 (scale of 0 to 4), an EASI score ≥16 (scale of 0 to 72), and a minimum BSA involvement of ≥10%. Eligible subjects enrolled into this trial had previous inadequate response to topical medication.
Subjects in the DUPIXENT group with baseline weight of <60 kg received an initial dose of 400 mg at Week 0, followed by 200 mg Q2W for 16 weeks. Subjects with baseline weight of ≥60 kg received an initial dose of 600 mg at Week 0, followed by 300 mg Q2W for 16 weeks. Subjects were permitted to receive rescue treatment at the discretion of the investigator. Subjects who received rescue treatment were considered non-responders.
In AD-1526, the mean age was 14.5 years, the median weight was 59.4 kg, 41% of subjects were female, 63% were White, 15% were Asian, and 12% were Black. At baseline 46% of subjects had an IGA score of 3 (moderate AD), 54% had an IGA score of 4 (severe AD), the mean BSA involvement was 57%, and 42% had received prior systemic immunosuppressants. Also, at baseline the mean EASI score was 36, and the weekly averaged Peak Pruritus NRS was 8 on a scale of 0-10. Overall, 92% of subjects had at least one co-morbid allergic condition; 66% had allergic rhinitis, 54% had asthma, and 61% had food allergies.
The primary endpoint was the proportion of subjects with an IGA 0 (clear) or 1 (almost clear) and at least a 2-point improvement from baseline to Week 16. Other evaluated outcomes included the proportion of subjects with EASI-75 or EASI-90 (improvement of at least 75% or 90% in EASI from baseline, respectively), and reduction in itch as measured by the Peak Pruritus NRS (≥4-point improvement).
The efficacy results at Week 16 for AD-1526 are presented in Table 3.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA greater proportion of subjects randomized to DUPIXENT achieved an improvement in the Peak Pruritus NRS compared to placebo (defined as >4-point improvement at Week 4). (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePediatric Subjects 6 to 11 Years of Age with Atopic Dermatitis: The efficacy and safety of DUPIXENT use concomitantly with TCS in pediatric subjects was evaluated in a multicenter, randomized, double-blind, placebo-controlled trial (AD-1652; NCT03345914) in 367 subjects 6 to 11 years of age, with AD defined by an IGA score of 4 (scale of 0 to 4), an EASI score ≥21 (scale of 0 to 72), and a minimum BSA involvement of ≥15%. Eligible subjects enrolled into this trial had previous inadequate response to topical medication. Enrollment was stratified by baseline weight (<30 kg; ≥30 kg).
Subjects in the DUPIXENT Q4W + TCS group received an initial dose of 600 mg on Day 1, followed by 300 mg Q4W from Week 4 to Week 12, regardless of weight. Subjects in the DUPIXENT Q2W + TCS group with baseline weight of <30 kg received an initial dose of 200 mg on Day 1, followed by 100 mg Q2W from Week 2 to Week 14, and subjects with baseline weight of ≥30 kg received an initial dose of 400 mg on Day 1, followed by 200 mg Q2W from Week 2 to Week 14. Subjects were permitted to receive rescue treatment at the discretion of the investigator. Subjects who received rescue treatment were considered non-responders.
In AD-1652, the mean age was 8.5 years, the median weight was 29.8 kg, 50% of subjects were female, 69% were White, 17% were Black, and 8% were Asian. At baseline, the mean BSA involvement was 58%, and 17% had received prior systemic non-steroidal immunosuppressants.
Also, at baseline the mean EASI score was 37.9, and the weekly average of daily worst itch score was 7.8 on a scale of 0-10. Overall, 92% of subjects had at least one co-morbid allergic condition; 64% had food allergies, 63% had other allergies, 60% had allergic rhinitis, and 47% had asthma.
The primary endpoint was the proportion of subjects with an IGA 0 (clear) or 1 (almost clear) at Week 16. Other evaluated outcomes included the proportion of subjects with EASI-75 or EASI- 90 (improvement of at least 75% or 90% in EASI from baseline, respectively), and reduction in itch as measured by the Peak Pruritus NRS (≥4-point improvement).
Table 4 presents the results by baseline weight strata for the approved dose regimens. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA greater proportion of subjects randomized to DUPIXENT + TCS achieved an improvement in the Peak Pruritus NRS compared to placebo + TCS (defined as ≥4-point improvement at Week 16). (See Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePediatric Subjects 6 Months to 5 Years of Age with Atopic Dermatitis: The efficacy and safety of DUPIXENT use concomitantly with TCS in pediatric subjects was evaluated in a multicenter, randomized, double-blind, placebo-controlled trial (AD-1539; NCT03346434) in 162 subjects 6 months to 5 years of age, with moderate-to-severe AD defined by an IGA score ≥3 (scale of 0 to 4), an EASI score ≥16 (scale of 0 to 72), and a minimum BSA involvement of ≥10%. Eligible subjects enrolled into this trial had previous inadequate response to topical medication. Enrollment was stratified by baseline weight (≥5 to <15 kg and ≥15 to <30 kg).
Subjects in the DUPIXENT Q4W + TCS group with baseline weight of ≥5 to <15 kg received an initial dose of 200 mg on Day 1, followed by 200 mg Q4W from Week 4 to Week 12, and subjects with baseline weight of ≥15 to <30 kg received an initial dose of 300 mg on Day 1, followed by 300 mg O4W from Week 4 to Week 12. Subjects were permitted to receive rescue treatment at the discretion of the investigator. Subjects who received rescue treatment were considered non-responders.
In AD-1539, the mean age was 3.8 years, the median weight was 16.5 kg, 39% of subjects were female, 69% were White, 19% were Black, and 6% were Asian. At baseline, the mean BSA involvement was 58%, and 29% of subjects had received prior systemic immunosuppressants. Also, at baseline the mean EASI score was 34.1, and the weekly average of daily worst scratch/itch score was 7.6 on a scale of 0-10. Overall, 81.4% of subjects had at least one co-morbid allergic condition; 68.3% had food allergies, 52.8% had other allergies, 44.1% had allergic rhinitis, and 25.5% had asthma.
The primary endpoint was the proportion of subjects with an IGA 0 (clear) or 1 (almost clear) at Week 16. Other evaluated outcomes included the proportion of subjects with EASI-75 or EASI 90 (improvement of at least 75% or 90% in EASI from baseline, respectively), and reduction in itch as measured by the Worst Scratch/Itch NRS (≥4-point improvement).
The efficacy results at Week 16 for AD-1539 are presented in Table 5. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAsthma: The asthma development program included three randomized, double-blind, placebo-controlled, parallel-group, multi-center studies (DRI12544, QUEST, and VENTURE) of 24 to 52 weeks in treatment duration which enrolled a total of 2888 patients (12 years of age and older).
Patients enrolled in DRI12544 and QUEST studies were required to have a history of 1 or more asthma exacerbations that required treatment with systemic corticosteroids or emergency department visit or hospitalization for the treatment of asthma in the year prior to study entry.
Patients enrolled in VENTURE study required dependence on daily oral corticosteroids in addition to regular use of high-dose inhaled corticosteroids plus an additional controller(s).
The effects of DUPIXENT treatment discontinuation on severe exacerbations and FEV1 were assessed in the DRI12544 study during the 16-week follow-up period. Patients in both the overall and the baseline blood eosinophil count of ≥300 cells/mcL populations experienced a gradual return to baseline asthma status, with no evidence of rebound effect.
In all 3 studies, patients were enrolled without requiring a minimum baseline blood eosinophil or other Type 2 biomarker (e.g. FeNO or IgE) level.
In the QUEST and VENTURE studies, patients with baseline blood eosinophil level of >1500 cells/mcL (<1.3%) were excluded.
DUPIXENT was administered as add-on to background asthma treatment.
Patients continued background asthma therapy throughout the duration of the studies except in VENTURE study in which OCS dose was tapered as described as follows.
DRI12544 study: DRI12544 was a 24-week dose-ranging study which included 776 patients (18 years of age and older). DUPIXENT compared with placebo was evaluated in adult patients with moderate-to-severe asthma on a medium- or-high dose inhaled corticosteroid and a long-acting beta agonist.
Patients were randomized to receive either 200 mg (N=150) or 300 mg (N=157); DUPIXENT every other week or 200 mg (N=154) or 300 mg (N=157); DUPIXENT every 4 weeks following an initial dose of 400 mg, 600 mg or placebo (N=158), respectively.
The primary endpoint was change from baseline to Week 12 in FEV1 (L). Other endpoints included percent change from baseline in FEV1 and annualized rate of severe asthma exacerbation events during the 24-week placebo-controlled treatment period.
Results were evaluated in the overall population and subgroups based on baseline blood eosinophil count (≥300 cells/mcL and <300 cells/mcL).
Additional secondary endpoints included mean change from baseline and responder rates in the patient reported Asthma Control Questionnaire (ACQ-5) and Asthma Quality of Life Questionnaire, Standardized Version (AQLQ(S)) scores.
EFC13579 (QUEST) study: QUEST was a 52-week study which included 1902 patients (12 years of age and older). DUPIXENT compared with placebo was evaluated in 107 pediatric subjects 12 to 17 years of age and 1795 adult patients with moderate-to-severe asthma on a medium- or high-dose inhaled corticosteroid (ICS) and a minimum of one and up to two controller medications.
Patients requiring a third controller were allowed to participate in this study. Patients were randomized to receive either 200 mg (N=631) or 300 mg (N=633) DUPIXENT every other week (or matching placebo for either 200 mg [N=317] or 300 mg [N=321] every other week) following an initial dose of 400 mg, 600 mg or placebo respectively.
The primary endpoints were the annualized rate of severe exacerbation events during the 52-week placebo-controlled period and change from baseline in pre-bronchodilator FEV1 at Week 12 in overall population (unrestricted by minimum baseline eosinophils or other Type 2 biomarkers).
Additional secondary endpoints included exacerbation rates and FEV1 in patients with different baseline levels of eosinophils as well as mean change from baseline and responder rates in theACQ-5 and AQLQ(S) scores.
EFC13691 (VENTURE) study: VENTURE was a 24-week oral corticosteroid-reduction study in 210 patients with asthma who required daily oral corticosteroids in addition to regular use of high-dose inhaled corticosteroids plus an additional controller.
After optimizing the OCS dose during the screening period, patients received 300 mg DUPIXENT (N=103) or placebo (N=107) once every other week for 24 weeks following an initial dose of 600 mg or placebo.
Patients continued to receive their existing asthma medicine during the study; however their OCS dose which was reduced every 4 weeks during the OCS reduction phase (Week 4-20), as long as asthma control was maintained. The OCS reduction was performed according to algorithm specified in the protocol.
The primary endpoint was the percent reduction of oral corticosteroid dose at Week 24 compared with the baseline dose, while maintaining asthma control in the overall population (unrestricted by minimum baseline eosinophils or other Type 2 biomarkers). The key secondary endpoints were the proportion of patients achieving a reduction of 50% or greater in their OCS dose compared with baseline and proportion of patients achieving a reduction of OCS dose to <5 mg/day at Week 24 while maintaining asthma control.
Additional secondary endpoints included the annualized rate of severe exacerbation events during treatment period and mean change from baseline and responder rate in the ACQ-5 and AQLQ(S) scores.
The demographics and baseline characteristics of these 3 studies are provided in Table 6 as follows. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageExacerbations: DRI12544, QUEST, and VENTURE studies evaluated the frequency of severe asthma exacerbations.
Exacerbations were defined as deterioration of asthma requiring the use of systemic corticosteroids for at least 3 days or hospitalization or emergency room visit due to asthma that required systemic corticosteroids.
For patients on maintenance corticosteroids, an asthma exacerbation was defined as a temporary increase in oral corticosteroid dose for at least 3 days.
In the overall population, patients receiving either DUPIXENT 200 mg or 300 mg every other week had significant reductions in the rate of severe asthma exacerbations compared to placebo (see Table 7).
In the pooled analysis of the DRI12544 and QUEST studies, the rate of severe exacerbations leading to hospitalizations and/or emergency room visits was reduced by 25.5% and 46.9% with DUPIXENT 200 mg or 300 mg every other week, respectively. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePrespecified subgroup analyses of DRI12544, QUEST, and VENTURE studies demonstrated that there were greater reductions in severe exacerbations in patients with higher baseline levels of markers for Type 2 inflammation such as eosinophil level and FeNO.
Prespecified subgroup analyses of DRI12544 and QUEST demonstrated that there were greater reductions in severe exacerbations in subjects with higher baseline blood eosinophil levels. In QUEST, reductions in exacerbations were significant in the subgroup of subjects with baseline blood eosinophils ≥150 cells/mcL. In subjects with baseline blood eosinophil count <150 cells/mcL, similar severe exacerbation rates were observed between DUPIXENT and placebo.
In all studies, when compared to placebo greater reductions in severe exacerbations were also seen in patients with baseline FeNO ≥25 ppb.
In the QUEST study, patients receiving medium-dose ICS showed a similar reduction in rate of severe asthma exacerbations compared to patients receiving high-dose ICS. (See Table 8 and Figure 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
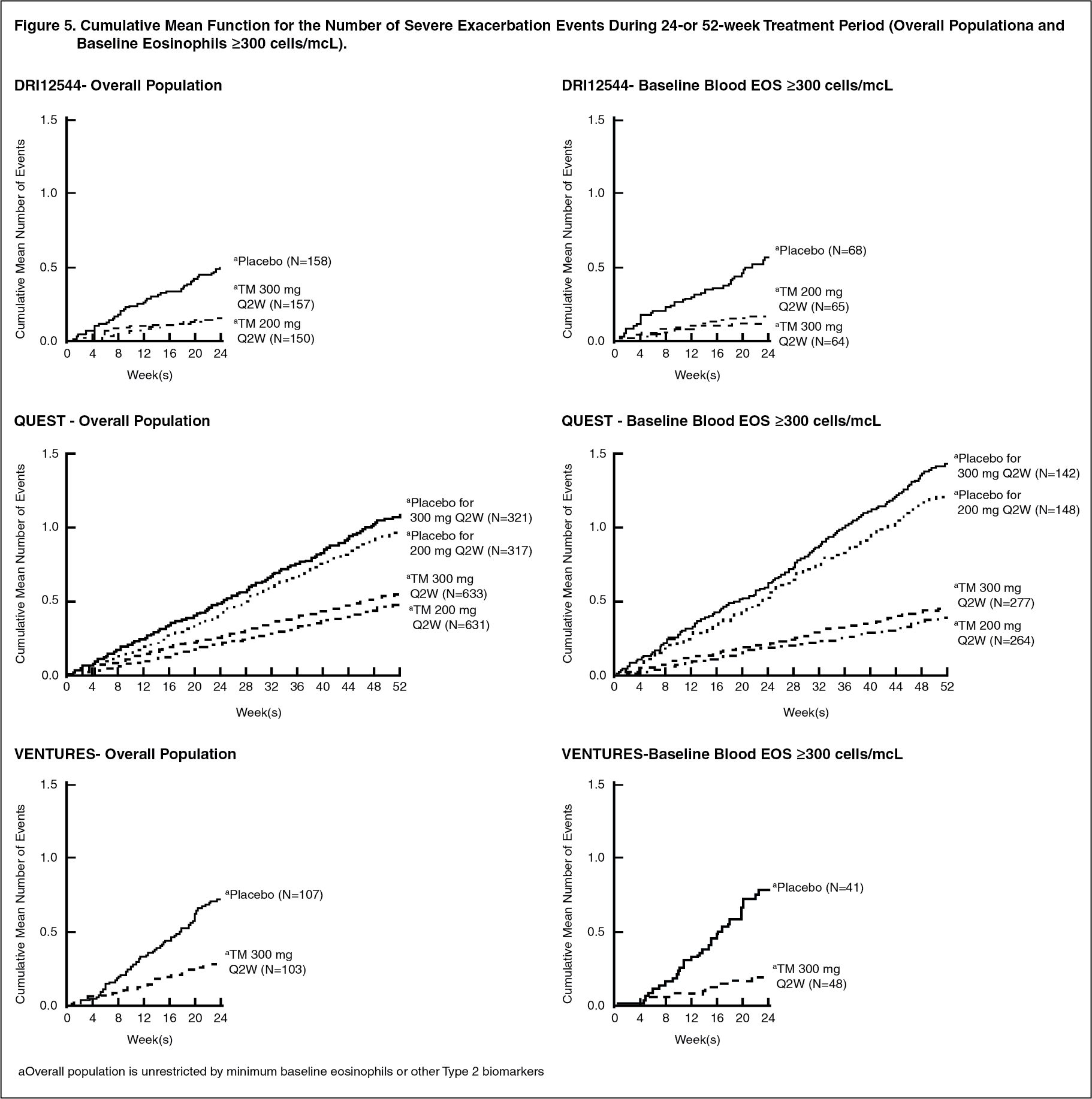
Click on icon to see table/diagram/imageThe cumulative mean number of severe exacerbation events in DRI12544, QUEST, and VENTURE studies (Overall Population and Baseline Eosinophils ≥300 cells/mcL) during the 24- or 52-week treatment period is shown in Figure 5. (See Figure 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOver the course of the studies, patients in both DUPIXENT dose groups had lower cumulative number of events compared with patients in their respective placebo groups.
Lung Function: Significant increases in pre-bronchodilator FEV1 were observed at week 12 for DRI12544 and QUEST trials in the primary analysis populations (subjects with baseline blood eosinophil count of ≥300 cells/mcL in DRI12544 and the overall population in the QUEST trial.
Subgroup analysis of DRI12544, QUEST, and VENTURE studies demonstrated that patients with baseline blood eosinophil count of ≥150 and ≥300 cells/mcL showed greater improvement in FEV1 compared with the overall population (Table 9).
Clinically meaningful improvements in FEV1 were observed in patients with baseline eosinophils <300 cell/mcL, although less than in the population with baseline blood eosinophil count ≥300 cells/mcL. Magnitude of effect was directly correlated with baseline eosinophil counts at all baseline eosinophil levels studied.
In the QUEST study, compared to placebo, greater improvements in FEV1 were also seen in patients with FeNO ≥25 and ≥50 ppb.
Improvement in FEV1 was similar whether patients were receiving medium dose ICS, high-dose ICS, or OCS. (See Tables 9, 10 and Figure 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSignificant improvements in FEV1 were observed as early as Week 2 (DRI12544, QUEST, and VENTURE) following the first dose of DUPIXENT for both the 200 mg and 300 mg dose strengths and were maintained through Week 24 (DRI12544 and VENTURE) and Week 52 (QUEST) (Figure 6). (See Figure 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdditional Secondary Endpoints: ACQ-5 and AQLQ(S) were analysed at both a cohort level (mean change from baseline) and an individual level (responder analyses) at 24 weeks (DRI12544) and at 52 weeks (QUEST).
The responder rate was defined as an improvement in score of 0.5 or more (scale range 0-6 for ACQ-5 and 1-7 for AQLQ(S)). Improvements in ACQ-5 and AQLQ(S) were observed as early as Week 2 and maintained for 24 weeks in DRI12544 study and 52 weeks in QUEST study.
Similar results were observed in the VENTURE study. In asthma patients with comorbid upper airway disease DUPIXENT treatment also reduced upper airway symptoms.
Patients with asthma and comorbid chronic rhinosinusitis (CRS) with or without nasal polyposis, and/or comorbid allergic rhinitis (AR), reported their health-related quality of life on disease specific questionnaires; the 22-Item Sino Nasal Outcome Test (SNOT-22) for CRS patients and Standardized Rhinoconjunctivitis Quality of Life Questionnaire (RQLQ (S)+ 12) for AR patients. Mean change from baseline in total scores on SNOT-22 and RQLQ (S)+ 12 were pre-specified endpoints in these subpopulations. Improvements in SNOT-22 and RQLQ (S)+ 12 total score were observed with DUPIXENT compared to placebo as early as week 12 and sustained over 52 weeks.
Oral Corticosteroid Reduction (VENTURE): The VENTURE study evaluated the effect of DUPIXENT on reducing the use of maintenance oral corticosteroids. The baseline mean oral corticosteroid use was 11.75 mg in the placebo group and 10.75 mg in the group receiving DUPIXENT.
Compared with placebo, patients receiving DUPIXENT achieved greater reductions in daily maintenance oral corticosteroid dose, while maintaining asthma control.
The results for primary and secondary endpoints of the VENTURE study are presented in Table 11. (See Table 11.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: The pharmacokinetics of dupilumab is similar in subjects with atopic dermatitis and asthma.
Absorption: Following an initial subcutaneous (SC) dose of 600 mg, 400 mg, or 300 mg dupilumab reached peak mean ± SD concentrations (Cmax) of 70.1±24.1 mcg/mL or 41.8±12.4 mcg/mL, or 30.5±9.39 mcg/mL respectively, by approximately 1 week post dose.
Steady-state concentrations were achieved by Week 16 following the administration of 600 mg starting dose and 300 mg dose either weekly (twice the recommended dosing frequency) or Q2W, or 400 mg starting dose and 200 mg dose Q2W, or 300 mg Q2W without loading dose. Across clinical trials, the mean ± SD steady-state trough concentrations ranged from 60.3±35.1 mcg/mL to 80.2±35.3 mcg/mL for 300 mg administered Q2W, from 173±75.9 mcg/mL to 193±77.0 mcg/mL for 300 mg administered weekly, and from 29.2±18.7 to 36.5±22.2 mg/L for 200 mg administered Q2W.
The bioavailability of dupilumab following a SC dose is estimated similar between AD and, asthma patients, ranging between 61% and 64%.
Distribution: The estimated total volume of distribution was approximately 4.8±1.3 L.
Elimination: The metabolic pathway of dupilumab has not been characterized. As a human monoclonal IgG4 antibody, dupilumab is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG.
After the last steady-state dose of 300 mg QW, 300 mg Q2W, 200 mg Q2W, 300 mg Q4W, or 200 mg Q4W dupilumab, the median times to non-detectable concentration (<78 ng/mL) are ranged from 9 to 13 weeks in adults and pediatric subjects 12 to 17 years of age. Population pharmacokinetic analyses indicate the median times to non-detectable concentration are approximately 1.5 times (up to 19 weeks) and 2.5 times (up to 32 weeks) longer in pediatric subjects 6 to 11 years of age and pediatric subjects 6 months to 5 years of age, respectively.
Dose Linearity: Dupilumab exhibited nonlinear target-mediated pharmacokinetics with exposures increasing in a greater than dose-proportional manner. The systemic exposure increased by 30-fold when the dose increased 8-fold following a single dose of dupilumab from 75 mg to 600 mg (i.e., 0.25-times to 2-times the recommended dose).
Weight: Dupilumab trough concentrations were lower in subjects with higher body weight.
Age: Based on population pharmacokinetic analysis, age did not affect dupilumab clearance in adults and in pediatric subjects 6 to 17 years of age. In pediatric subjects from 6 months to 5 years of age, clearance increased with age.
Immunogenicity: Development of antibodies to dupilumab was associated with lower serum dupilumab concentrations. A few subjects who had high antibody titers also had no detectable serum dupilumab concentrations.
Specific Populations: Geriatric Patients: In subjects who are 65 years and older, the mean ± SD steady-state trough concentrations of dupilumab were 69.4±31.4 mcg/mL and 166±62.3 mcg/mL, respectively, for 300 mg administered Q2W and weekly, and 39.7±21.7 mcg/mL for 200 mg administered Q2W.
Pediatric Patients: Atopic Dermatitis: For pediatric subjects 12 to 17 years of age with atopic dermatitis receiving every other week dosing (Q2W) with either 200 mg (<60 kg) or 300 mg (≥60 kg), the mean ± SD steady-state trough concentration of dupilumab was 54.5±27.0 mcg/mL.
For pediatric subjects 6 to 11 years of age with atopic dermatitis receiving every other week dosing (Q2W) with 200 mg (≥30 kg) or every four week dosing (Q4W) with 300 mg (<30 kg), mean ± SD steady-state trough concentration was 86.0±34.6 mcg/mL and 98.7±33.2 mcg/mL, respectively.
For pediatric subjects 6 months to 5 years of age with atopic dermatitis receiving every four week dosing (Q4W) with 300 mg (≥15 to <30 kg) or 200 mg (≥5 to <15 kg), the mean ± SD steady-state trough concentration was 110±42.8 mcg/mL and 109±50.8 mcg/mL, respectively.
Asthma: A total of 107 pediatric subjects aged 12 to 17 years with asthma were enrolled in QUEST. The mean ± SD steady-state trough concentrations of dupilumab were 107±51.6 mcg/mL and 46.7±26.9 mcg/mL, respectively, for 300 mg or 200 mg administered Q2W.
Renal or Hepatic Impairment: No formal trial of the effect of hepatic or renal impairment on the pharmacokinetics of dupilumab was conducted.
Drug Interaction Studies: An effect of dupilumab on the PK of co-administered medications is not expected. Based on the population analysis, commonly co-administered medications had no effect on DUPIXENT pharmacokinetics in patients with moderate-to-severe asthma.
Cytochrome P450 Substrates: The effects of dupilumab on the pharmacokinetics of midazolam (metabolized by CYP3A4), warfarin (metabolized by CYP2C9), omeprazole (metabolized by CYP2C19), metoprolol (metabolized by CYP2D6), and caffeine (metabolized by CYP1A2) were evaluated in a study with 12-13 evaluable subjects with atopic dermatitis (a SC loading dose of 600 mg followed by 300 mg SC weekly for six weeks). No clinically significant changes in AUC were observed. The largest effect was observed for metoprolol (CYP2D6) with an increase in AUC of 29%.
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Animal studies have not been conducted to evaluate the carcinogenic or mutagenic potential of dupilumab.
No effects on fertility parameters such as reproductive organs, menstrual cycle length, or sperm analysis were observed in sexually mature mice that were subcutaneously administered a homologous antibody against IL-4Rα at doses up to 200 mg/kg/week.