Each capsule contains 30 mg of duloxetine (as hydrochloride).
Each capsule contains 60 mg of duloxetine (as hydrochloride).
Excipients/Inactive Ingredients: Excipient(s) with known effect 30 mg: each capsule may contain up to 56 mg sucrose.
Excipient(s) with known effect 60 mg: each capsule may contain up to 111 mg sucrose.
Capsule content: Hypromellose, Hypromellose acetate succinate, Sucrose, Sugar spheres, Talc, Titanium dioxide (E171), Triethyl citrate.
Capsule shell: 30 mg: Gelatin, Sodium Lauryl Sulfate, Titanium Dioxide (E171), Indigo Carmine (E132), Edible Green Ink. Edible Green Ink contains: Black Iron Oxide-Synthetic (E172), Yellow Iron Oxide- Synthetic (E172), Propylene glycol, Shellac.
60 mg: Gelatin, Sodium Lauryl Sulfate, Titanium Dioxide (E171), Indigo Carmine (E132), Yellow Iron Oxide (E172), Edible White Ink. Edible White Ink contains: Titanium Dioxide (E171), Propylene glycol, Shellac, Povidone.
Pharmacotherapeutic group: Other antidepressants. ATC code: N06AX2.
Pharmacology: Pharmacodynamics: Mechanism of action: Duloxetine is a combined serotonin (5-HT) and noradrenaline (NA) reuptake inhibitor. It weakly inhibits dopamine reuptake with no significant affinity for histaminergic, dopaminergic, cholinergic and adrenergic receptors. Duloxetine dose-dependently increases extracellular levels of serotonin and noradrenaline in various brain areas of animals.
Pharmacodynamic effects: Duloxetine normalized pain thresholds in several preclinical models of neuropathic and inflammatory pain and attenuated pain behavior in a model of persistent pain. The pain inhibitory action of duloxetine is believed to be result of potentiation of descending inhibitory pain pathways within the central nervous system.
Clinical efficacy and safety: Major Depressive Disorder: Cymbalta was studied in a clinical programme involving 3,158 patients (1,285 patient-years of exposure) meeting DSM-IV criteria for major depression. The efficacy of Cymbalta at the recommended dose of 60 mg once a day was demonstrated in three out of three randomized, double-blind, placebo-controlled, fixed dose acute studies in adult outpatients with major depressive disorder. Overall, Cymbalta's efficacy has been demonstrated at daily doses between 60 and 120 mg in a total of five out of seven randomized, double-blind, placebo-controlled, fixed dose acute studies in adult outpatients with major depressive disorder.
Cymbalta demonstrated statistical superiority over placebo as measured by improvement in the 17-item Hamilton Depression Rating Scale (HAM-D) total score (including both the emotional and somatic symptoms of depression). Response and remission rates were also statistically significantly higher with Cymbalta compared with placebo. Only a small proportion of patients included in pivotal clinical trials had severe depression (baseline HAM-D>25).
In a relapse prevention study, patients responding to 12-weeks of acute treatment with open-label Cymbalta 60 mg once daily were randomised to either Cymbalta 60 mg once daily or placebo for a further 6-months. Cymbalta 60 mg once daily demonstrated a statistically significant superiority compared to placebo (p=0.004) on the primary outcome measure, the prevention of depressive relapse, as measured by time to relapse. The incidence of relapse during the 6-months double-blind follow-up period was 17% and 29% for duloxetine and placebo, respectively.
The effect of Cymbalta 60 mg once a day in elderly depressed patients (≥65 years) was specifically examined in a study that showed a statistically significant difference in the reduction of the HAMD17 score for duloxetine-treated patients compared to placebo. Tolerability of Cymbalta 60 mg once a day in elderly patients was comparable to that seen in the younger adults. However, data on elderly patients exposed to the maximum dose (120 mg per day) are limited and thus, caution is recommended when treating this population.
Generalised Anxiety Disorder: Cymbalta demonstrated statistically significant superiority over placebo in five out of five studies including four randomised, double-blind, placebo-controlled acute studies and a relapse prevention study in adult patients with generalised anxiety disorder.
Cymbalta demonstrated statistically significant superiority over placebo as measured by improvement in the Hamilton Anxiety Scale (HAM-A) total score and by the Sheehan Disability Scale (SDS) global functional impairment score. Response and remission rates were also higher with Cymbalta compared to placebo. Cymbalta showed comparable efficacy results to venlafaxine in terms of improvements on the HAM-A total score.
In a relapse prevention study, patients responding to 6 months of acute treatment with open-label Cymbalta were randomised to either Cymbalta or placebo for further 6-months. CYMBALTA 60 mg to 120 mg once daily demonstrated statistically significant superiority compared to placebo (p<0.001) on the prevention of relapse, as measured by time to relapse. The incidence of relapse during the 6-months double-blind follow-up period was 14% for Cymbalta and 42% for placebo.
The efficacy of Cymbalta 30-120 mg (flexible dosing) once a day in elderly patients (>65 years) with generalised anxiety disorder was evaluated in a study that demonstrated statistically significant improvement in the HAM-A total score for duloxetine-treated patients compared to placebo-treated patients. The efficacy and safety of Cymbalta 30-120 mg once daily in elderly patients with generalised anxiety disorder was similar to that seen in studies of younger adult patients. However, data on elderly patients exposed to the maximum dose (120 mg per day) are limited and, thus, caution is recommended when using this dose with the elderly population.
Diabetic Peripheral Neuropathic Pain: The efficacy of Cymbalta as a treatment for diabetic neuropathic pain was established in 2 randomised, 12-week, double-blind, placebo-controlled, fixed dose studies in adults (22 to 88 years) having diabetic neuropathic pain for at least 6 months. Patients meeting diagnostic criteria for major depressive disorder were excluded from these trials. The primary outcome measure was the weekly mean of 24-hour average pain, which was collected in a daily diary by patients on an 11-point Likert scale.
In both studies, Cymbalta 60 mg once daily and 60 mg twice daily significantly reduced pain compared with placebo. The effect in some patients was apparent in the first week of treatment.
The difference in mean improvement between the two active treatment arms was not significant. At least 30% reported pain reduction was recorded in approximately 65% of duloxetine treated patients versus 40% for placebo. The corresponding figures for at least 50% pain reduction were 50% and 26% respectively. Clinical response rates (50% or greater improvement in pain) were analysed according to whether or not the patient experienced somnolence during treatment. For patients not experiencing somnolence, clinical response was observed in 47% of patients receiving duloxetine and 27% patients on placebo. Clinical response rates in patients experiencing somnolence were 60% on duloxetine and 30% on placebo. Patients not demonstrating a pain reduction of 30% within 60 days of treatment were unlikely to reach this level during further treatment.
In an open label long-term uncontrolled study, the pain reduction in patients responding to 8-weeks of acute treatment of Cymbalta 60 mg once daily was maintained for a further 6-months as measured by change on the Brief Pain Inventory (BPI) 24-hour average pain item.
Paediatric population: Duloxetine has not been studied in patients under the age of 7. Two randomized, double-blind, parallel clinical trials were performed in 800 paediatric patients aged 7 to 17 years with major depressive disorder (see Dosage & Administration). These two studies included a 10 week placebo and active (fluoxetine) controlled acute phase followed by six months period of active controlled extension treatment. Neither duloxetine (30-120 mg) nor the active control arm (fluoxetine 20-40 mg) statistically separated from placebo on change from baseline to endpoint in the Children's Depression Rating Scale-Revised (CDRS-R) total score. Discontinuation due to adverse events was higher in patients taking duloxetine compared with those treated with fluoxetine, mostly due to nausea. During the 10-week acute treatment period, suicidal behaviours were reported (duloxetine 0/333 [0%], fluoxetine 2/225 [0.9%], placebo 1/220 [0.5%]). Over the entire 36-week course of the study, 6 out of 333 patients initially randomized to duloxetine and 3 out of 225 patients initially randomized to fluoxetine experienced suicidal behaviour (exposure adjusted incidence 0.039 events per patient year for duloxetine, and 0.026 for fluoxetine). In addition, one patient who transitioned from placebo to duloxetine experienced a suicidal behaviour while taking duloxetine.
A randomised, double-blind, placebo-controlled study was performed in 272 patients aged 7–17 years with generalised anxiety disorder. The study included a 10 week placebo-controlled acute phase, followed by an 18 week extension treatment period. A flexible dose regimen was used in this study, to allow for slow dose escalation from 30 mg once daily to higher doses (maximum 120 mg once daily). Treatment with duloxetine showed a statistically significantly greater improvement in GAD symptoms, as measured by PARS severity score for GAD (mean difference between duloxetine and placebo of 2.7 points [95% CI 1.3-4.0]), after 10 weeks of treatment. The maintenance of the effect has not been evaluated. There was no statistically significant difference in discontinuation due to adverse events between duloxetine and placebo groups during the 10-week acute treatment phase. Two patients who transitioned from placebo to duloxetine after the acute phase experienced suicidal behaviours while taking duloxetine during the extension phase. A conclusion on the overall benefit/risk in this age group has not been established (see Dosage & Administration and Adverse Reactions).
Pharmacokinetics: Duloxetine is administered as a single enantiomer. Duloxetine is extensively metabolised by oxidative enzymes (CYP1A2 and the polymorphic CYP2D6), followed by conjugation. The pharmacokinetics of duloxetine demonstrate large intersubject variability (generally 50-60%), partly due to gender, age, smoking status and CYP2D6 metaboliser status.
Absorption: Duloxetine is well absorbed after oral administration with a Cmax occurring 6 hours post dose. The absolute oral bioavailability of duloxetine ranged from 32% to 80% (mean of 50%). Food delays the time to reach the peak concentration from 6 to 10 hours and it marginally decreases the extent of absorption (approximately 11%). These changes do not have any clinical significance.
Distribution: Duloxetine is approximately 96% bound to human plasma proteins. Duloxetine binds to both albumin and alpha1-acid glycoprotein. Protein binding is not affected by renal or hepatic impairment.
Biotransformation: Duloxetine is extensively metabolised and the metabolites are excreted principally in urine. Both cytochromes P450-2D6 and 1A2 catalyse the formation of the two major metabolites glucuronide conjugate of 4-hydroxy duloxetine and sulphate conjugate of 5hydroxy, 6-methoxy duloxetine. Based upon in vitro studies, the circulating metabolites of duloxetine are considered pharmacologically inactive. The pharmacokinetics of duloxetine in patients who are poor metabolisers with respect to CYP2D6 has not been specifically investigated. Limited data suggest that the plasma levels of duloxetine are higher in these patients.
Elimination: The elimination half-life of duloxetine ranges from 8 to 17 hours (mean of 12 hours). After an intravenous dose the plasma clearance of duloxetine ranges from 22 l/hr to 46 l/hr (mean of 36 l/hr). After an oral dose the apparent plasma clearance of duloxetine ranges from 33 to 261 l/hr (mean 101 l/hr).
Special Populations: Gender: Pharmacokinetic differences have been identified between males and females (apparent plasma clearance is approximatively 50% lower in females). Based upon the overlap in the range of clearance, gender-based pharmacokinetic differences do not justify the recommendation for using a lower dose for female patients.
Age: Pharmacokinetic differences have been identified between younger and elderly females (≥65 years) (AUC increases by about 25% and half-life is about 25% longer in the elderly), although the magnitude of these changes is not sufficient to justify adjustments to the dose. As a general recommendation, caution should be exercised when treating the elderly (see Dosage & Administration and Precautions).
Renal impairment: End stage renal disease (ESRD) patients receiving dialysis had 2-fold higher duloxetine Cmax and AUC values compared with healthy subjects. Pharmacokinetic data on duloxetine is limited in patients with mild or moderate renal impairment.
Hepatic impairment: Moderate liver disease (Child-Pugh Class B) affected the pharmacokinetics of duloxetine. Compared with healthy subjects, the apparent plasma clearance of duloxetine was 79% lower, the apparent terminal half-life was 2.3 times longer, and the AUC was 3.7 times higher in patients with moderate liver disease. The pharmacokinetics of duloxetine and its metabolites have not been studied in patients with mild or severe hepatic insufficiency.
Breast-feeding mothers: The disposition of duloxetine was studied in 6 lactating women who were at least 12-week postpartum. Duloxetine is detected in breast milk, and steady-state concentrations in breastmilk are about one-fourth those in plasma. The amount of duloxetine in breast milk is approximately 7 μg/day while on 40 mg twice daily dosing. Lactation did not influence duloxetine pharmacokinetics.
Paediatric population: Pharmacokinetics of duloxetine in paediatric patients aged 7 to 17 years with major depressive disorder following oral administration of 20 to 120 mg once daily dosing regimen was characterized using population modelling analyses based on data from 3 studies.
The model-predicted duloxetine steady state plasma concentrations in paediatric patients were mostly within the concentration range observed in adult patients.
Toxicology: Preclinical safety data: Duloxetine was not genotoxic in a standard battery of tests and was not carcinogenic in rats.
Multinucleated cells were seen in the liver in the absence of other histopathological changes in the rat carcinogenicity study. The underlying mechanism and the clinical relevance are unknown. Female mice receiving duloxetine for 2 years had an increased incidence of hepatocellular adenomas and carcinomas at the high dose only (144 mg/kg/day), but these were considered to be secondary to hepatic microsomal enzyme induction. The relevance of this mouse data to humans is unknown. Female rats receiving duloxetine (45 mg/kg/day) before and during mating and early pregnancy had a decrease in maternal food consumption and body weight, oestrous cycle disruption, decreased live birth indices and progeny survival, and progeny growth retardation at systemic exposure levels estimated to be at the most at maximum clinical exposure (AUC). In an embryotoxicity study in the rabbit, a higher incidence of cardiovascular and skeletal malformations was observed at systemic exposure levels below the maximum clinical exposure (AUC). No malformations were observed in another study testing a higher dose of a different salt of duloxetine. In prenatal/postnatal toxicity studies in the rat, duloxetine induced adverse behavioural effects in the offspring at exposures below maximum clinical exposure (AUC).
Studies in juvenile rats reveal transient effects on neurobehaviour, as well as significantly decreased body weight and food consumption; hepatic enzyme induction; and hepatocellular vacuolation at 45 mg/kg/day. The general toxicity profile of duloxetine in juvenile rats was similar to that in adult rats. The no-adverse effect level was determined to be 20 mg/kg/day.
Treatment of major depressive disorder.
Management of neuropathic pain associated with diabetic peripheral neuropathy in adults.
Treatment of generalized anxiety disorder.
Posology: Major depressive disorder: The starting and recommended maintenance dose is 60 mg once daily with or without food.
Dosages above 60 mg once daily, up to a maximum dose of 120 mg per day have been evaluated from a safety perspective in clinical trials. However, there is no clinical evidence suggesting that patients not responding to the initial recommended dose may benefit from dose up-titrations.
Therapeutic response is usually seen after 2-4 weeks of treatment.
After consolidation of the antidepressive response, it is recommended to continue treatment for several months in order to avoid relapse. In patients responding to duloxetine, and with a history of repeated episodes of major depression, further long-term treatment at a dose of 60 to 120 mg/day could be considered.
Diabetic peripheral neuropathic pain: The starting and recommended maintenance dose is 60 mg daily with or without food. Dosages above 60 mg once daily, up to a maximum dose of 120 mg per day administered in evenly divided doses, have been evaluated from a safety perspective in clinical trials. The plasma concentration of duloxetine displays large inter-individual variability (see Pharmacology: Pharmacokinetics under Actions). Hence, some patients that respond insufficiently to 60 mg may benefit from a higher dose.
Response to treatment should be evaluated after 2 months. In patients with inadequate initial response, additional response after this time is unlikely.
The therapeutic benefit should be reassessed regularly (at least every three months) (see Pharmacology: Pharmacodynamics under Actions).
Generalized anxiety disorder: The recommended starting dose in patients with generalised anxiety disorder is 30 mg once daily with or without food. In patients with insufficient response the dose should be increased to 60 mg, which is the usual maintenance dose in most patients.
In patients with co-morbid major depressive disorder, the starting and maintenance dose is 60 mg once daily (please see also dosing recommendation above).
Doses up to 120 mg per day have been shown to be efficacious and have been evaluated from a safety perspective in clinical trials. In patients with insufficient response to 60 mg, escalation up to 90 mg or 120 mg may therefore be considered. Dose escalation should be based upon clinical response and tolerability.
After consolidation of the response, it is recommended to continue treatment for several months, in order to avoid relapse.
Special Populations: Elderly: No dosage adjustment is recommended for elderly patients solely on the basis of age. However, as with any medicine, caution should be exercised when treating the elderly, especially with Cymbalta 120 mg per day for major depressive disorder or generalised anxiety disorder, for which data are limited (see Pharmacology: Pharmacokinetics under Actions and Precautions).
Hepatic impairment: Cymbalta must not be used in patients with liver disease resulting in hepatic impairment (see Pharmacology: Pharmacokinetics under Actions and Contraindications).
Renal impairment: No dosage adjustment is necessary for patients with mild or moderate renal dysfunction (creatinine clearance 30 to 80 ml/min). Cymbalta must not be used in patients with severe renal impairment (creatinine clearance <30 ml/min; see Contraindications).
Paediatric population: Duloxetine should not be used in children and adolescents under the age of 18 years for the treatment of major depressive disorder because of safety and efficacy concerns (see Pharmacology: Pharmacodynamics under Actions, Precautions and Adverse Reactions).
The safety and efficacy of duloxetine for the treatment of generalised anxiety disorder in paediatric patients aged 7-17 years have not been established. Current available data are described in Pharmacology: Pharmacodynamics and Pharmacokinetics under Actions and Adverse reactions.
The safety and efficacy of duloxetine for the treatment of diabetic peripheral neuropathic pain has not been studied. No data are available.
Discontinuation of treatment: Abrupt discontinuation should be avoided. When stopping treatment with Cymbalta the dose should be gradually reduced over a period of at least one to two weeks in order to reduce the risk of withdrawal reactions (see Precautions and Adverse Reactions). If intolerable symptoms occur following a decrease in the dose or upon discontinuation of treatment, then resuming the previously prescribed dose may be considered. Subsequently, the physician may continue decreasing the dose, but at a more gradual rate.
Method of administration: For oral use.
Cases of overdoses, alone or in combination with other medicinal products, with duloxetine doses of 5400 mg were reported. Some fatalities have occurred, primarily with mixed overdoses, but also with duloxetine alone at a dose of approximately 1000 mg. Signs and symptoms of overdose (duloxetine alone or in combination with other medicinal products) included somnolence, coma, serotonin syndrome, seizures, vomiting and tachycardia.
No specific antidote is known for duloxetine, but if serotonin syndrome ensues, specific treatment (such as with cyproheptadine and/or temperature control) may be considered. A free airway should be established. Monitoring of cardiac and vital signs is recommended, along with appropriate symptomatic and supportive measures. Gastric lavage may be indicated if performed soon after ingestion or in symptomatic patients. Activated charcoal may be useful in limiting absorption. Duloxetine has a large volume of distribution and forced diuresis, haemoperfusion, and exchange perfusion are unlikely to be beneficial.
Hypersensitivity to active substance or to any of the excipients.
Concomitant use of Cymbalta with nonselective, irreversible monoamine oxidase inhibitors (MAOIs) is contraindicated (see Interactions).
Liver disease resulting in hepatic impairment (see Pharmacology: Pharmacokinetics under Actions).
Cymbalta should not be used in combination with fluvoxamine, ciprofloxacin or enoxacine (i.e. potent CYP1A2 inhibitors) since the combination results in elevated plasma concentrations of duloxetine (see Interactions).
Severe renal impairment (creatine clearance <30 mL/min) (see Precautions).
The initiation of treatment with Cymbalta is contraindicated in patients with uncontrolled hypertension that could expose patients to a potential risk of hypertensive crisis (see Precautions and Adverse Reactions).
Mania and seizures: Cymbalta should be used with caution in patients with a history of mania or a diagnosis of bipolar disorder, and/or seizures.
Mydriasis: Mydriasis has been reported in association with duloxetine, therefore, caution should be used when prescribing Cymbalta to patients with increased intraocular pressure, or those at risk of acute narrow-angle glaucoma.
Blood pressure and heart rate: Duloxetine has been associated with an increase in blood pressure and clinically significant hypertension in some patients. This may be due to the noradrenergic effect of duloxetine. Cases of hypertensive crisis have been reported with duloxetine, especially in patients with pre-existing hypertension. Therefore, in patients with known hypertension and/or other cardiac disease, blood pressure monitoring is recommended, especially during the first month of treatment. Duloxetine should be used with caution in patients whose conditions could be compromised by an increased heart rate or by an increase in blood pressure. Caution should also be exercised when duloxetine is used with medicinal products that may impair its metabolism (see Interactions). For patients who experience a sustained increase in blood pressure while receiving duloxetine either dose reduction or gradual discontinuation should be considered (see Adverse Reactions). In patients with uncontrolled hypertension duloxetine should not be initiated (see Contraindications).
Renal impairment: Increased plasma concentrations of duloxetine occur in patients with severe renal impairment on haemodialysis (creatinine clearance <30 ml/min). For patients with severe renal impairment, see Dosage & Administration and Contraindications for information on patients with mild or moderate renal dysfunction.
Serotonin syndrome: As with other serotonergic agents, serotonin syndrome, a potentially life-threatening condition, may occur with duloxetine treatment, particularly with concomitant use of other serotonergic agents (including SSRIs, SNRIs tricyclic antidepressants or triptans), with agents that impair metabolism of serotonin such as MAOIs, or with antipsychotics or other dopamine antagonists that may affect the serotonergic neurotransmitter systems (see Contraindications and Interactions).
Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g. tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination) and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhoea).
If concomitant treatment with duloxetine and other serotonergic agents that may affect the serotonergic and/or dopaminergic neurotransmitter systems is clinically warranted, careful observation of the patient is advised, particularly during treatment initiation and dose increases.
St John's wort: Adverse reactions may be more common during concomitant use of Cymbalta and herbal preparations containing St John's wort (Hypericum perforatum).
Suicide: Major Depressive Episodes and Generalised Anxiety Disorder: Depression is associated with an increased risk of suicidal thoughts, self harm and suicide (suicide-related events). This risk persists until significant remission occurs. As improvement may not occur during the first few weeks or more of treatment, patients should be closely monitored until such improvement occurs. It is general clinical experience that the risk of suicide may increase in the early stages of recovery.
Other psychiatric conditions for which Cymbalta is prescribed can also be associated with an increased risk of suicide-related events. In addition, these conditions may be co-morbid with major depressive disorder. The same precautions observed when treating patients with major depressive disorder should therefore be observed when treating patients with other psychiatric disorders.
Patients with a history of suicide-related events or those exhibiting a significant degree of suicidal thoughts prior to commencement of treatment are known to be at greater risk of suicidal thoughts or suicidal behaviour, and should receive careful monitoring during treatment. A meta-analysis of placebo-controlled clinical trials of antidepressant medicinal products in psychiatric disorders showed an increased risk of suicidal behaviour with antidepressants compared to placebo in patients less than 25 years old.
Cases of suicidal thoughts and suicidal behaviours have been reported during duloxetine therapy or early after treatment discontinuation (see Adverse Reactions).
Close supervision of patients and in particular those at high risk should accompany medicinal product therapy especially in early treatment and following dose changes. Patients (and caregivers of patients) should be alerted about the need to monitor for any clinical worsening, suicidal behaviour or thoughts and unusual changes in behaviour and to seek medical advice immediately if these symptoms present.
Diabetic Peripheral Neuropathic Pain: As with other medicinal products with similar pharmacological action (antidepressants), isolated cases of suicidal ideation and suicidal behaviours have been reported during duloxetine therapy or early after treatment discontinuation. Concerning risk factors for suicidality in depression, see above. Physicians should encourage patients to report any distressing thoughts or feelings at any time.
Use in children and adolescents under 18 years of age: Cymbalta should not be used in the treatment of children and adolescents under the age of 18 years. Suicide-related behaviours (suicide attempts and suicidal thoughts) and hostility (predominantly aggression, oppositional behaviour and anger) were more frequently observed in clinical trials among children and adolescents treated with antidepressants compared to those treated with placebo. If, based on clinical need, a decision to treat is nevertheless taken, the patient should be carefully monitored for the appearance of suicidal symptoms (see Pharmacology: Pharmacodynamics under Actions). In addition, long-term safety data in children and adolescents concerning growth, maturation, and cognitive and behavioural development are lacking (see Adverse Reactions).
Haemorrhage: There have been reports of bleeding abnormalities, such as ecchymoses, purpura and gastrointestinal haemorrhage with selective serotonin reuptake inhibitors (SSRIs) and serotonin/noradrenaline reuptake inhibitors (SNRIs), including duloxetine. Caution is advised in patients taking anticoagulants and/or medicinal products known to affect platelet function (e.g. NSAIDs or acetylsalicylic acid (ASA)), and in patients with known bleeding tendencies.
Hyponatraemia: Hyponatraemia has been reported when administering Cymbalta, including cases with serum sodium lower than 110 mmol/l. Hyponatraemia may be due to a syndrome of inappropriate anti-diuretic hormone secretion (SIADH). The majority of cases of hyponatraemia were reported in the elderly, especially when coupled with a recent history of, or condition pre-disposing to, altered fluid balance. Caution is required in patients at increased risk for hyponatraemia; such as elderly, cirrhotic, or dehydrated patients or patients treated with diuretics.
Discontinuation of treatment: Withdrawal symptoms when treatment is discontinued are common, particularly if discontinuation is abrupt (see Adverse Reactions). In clinical trials adverse events seen on abrupt treatment discontinuation occurred in approximately 45% of patients treated with Cymbalta and 23% of patients taking placebo. The risk of withdrawal symptoms seen with SSRI's and SNRI's may be dependent on several factors including the duration and dose of therapy and the rate of dose reduction. The most commonly reported reactions are listed in adverse reactions. Generally these symptoms are mild to moderate, however, in some patients they may be severe in intensity. They usually occur within the first few days of discontinuing treatment, but there have been very rare reports of such symptoms inpatients who have inadvertently missed a dose. Generally these symptoms are self-limiting and usually resolve within 2 weeks, though in some individuals they may be prolonged (2-3 months or more). It is therefore advised that duloxetine should be gradually tapered when discontinuing treatment over a period of no less than 2 weeks, according to the patient's needs (see Dosage & Administration).
Elderly: Data on the use of Cymbalta 120 mg in elderly patients with major depressive disorders are limited. Therefore, caution should be exercised when treating the elderly with the maximum dosage (see Pharmacology: Pharmacokinetics under Actions and Dosage & Administration).
Akathisia/psychomotor restlessness: The use of duloxetine has been associated with the development of akathisia, characterised by a subjectively unpleasant or distressing restlessness and need to move often accompanied by an inability to sit or stand still. This is most likely to occur within the first few weeks of treatment. In patients who develop these symptoms, increasing the dose may be detrimental.
Medicinal products containing duloxetine: Duloxetine is used under different trademarks in several indications (treatment of diabetic neuropathic pain, major depressive episodes, generalised anxiety disorder and stress urinary incontinence). The use of more than one of these products concomitantly should be avoided.
Hepatitis/increased liver enzymes: Cases of liver injury, including severe elevations of liver enzymes (>10 times upper limit of normal), hepatitis and jaundice have been reported with duloxetine (see Adverse Reactions). Most of them occurred during the first months of treatment. The pattern of liver damage was predominantly hepatocellular. Duloxetine should be used with caution in patients treated with other medicinal products associated with hepatic injury.
Sucrose: Cymbalta hard gastro-resistant capsules contain sucrose. Patients with rare hereditary problems of fructose intolerance, glucose-galactose malabsorption or sucrose-isomaltase insufficiency should not take this medicine.
Effects on ability to drive and use machines: No studies on the effects on the ability to drive and use machines have been performed. Cymbalta may be associated with sedation and dizziness. Patients should be instructed that if they experience sedation or dizziness they should avoid potentially hazardous tasks such as driving or operating machinery.
Fertility: Duloxetine had no effect on male fertility, and effects in females were only evident at doses that caused maternal toxicity.
Pregnancy: There are no adequate data on the use of duloxetine in pregnant women. Studies in animals have shown reproductive toxicity at systemic exposure levels (AUC) of duloxetine lower than the maximum clinical exposure (see Pharmacology: Toxicology: Preclinical safety data under Actions).
The potential risk for humans is unknown. Epidemiological data have suggested that the use of SSRIs in pregnancy, particularly in late pregnancy, may increase the risk of persistent pulmonary hypertension in the newborn (PPHN). Although no studies have investigated the association of PPHN to SNRI treatment, this potential risk cannot be ruled out with duloxetine taking into account the related mechanism of action (inhibition of the re-uptake of serotonin).
As with other serotonergic medicinal products, discontinuation symptoms may occur in the neonate after maternal duloxetine use near term. Discontinuation symptoms seen with duloxetine may include hypotonia, tremor, jitteriness, feeding difficulty, respiratory distress and seizures. The majority of cases have occurred either at birth or within a few days of birth.
Cymbalta should be used in pregnancy only if the potential benefit justifies the potential risk to the foetus. Women should be advised to notify their physician if they become pregnant, or intend to become pregnant, during therapy.
Breast feeding: Duloxetine is very weakly excreted into human milk, based on a study of 6 lactating patients, who did not breast feed their children. The estimated daily infant dose on a mg/kg basis is approximately 0.14% of the maternal dose (see Pharmacology: Pharmacokinetics under Actions). As the safety of duloxetine in infants is not known, the use of Cymbalta while breast-feeding is not recommended.
Summary of the safety profile: The most commonly reported adverse reactions in patients treated with Cymbalta were nausea, headache, dry mouth, somnolence, and dizziness. However, the majority of common adverse reactions were mild to moderate, they usually started early in therapy, and most tended to subside even as therapy was continued.
Tabulated summary of adverse reactions: The table gives the adverse reactions observed from spontaneous reporting and in placebo-controlled clinical trials.
Table: Adverse reactions: Frequency estimate: Very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to<1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. (See Table A and Table B.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Description of selected adverse reactions:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Description of selected adverse reactions: Discontinuation of duloxetine (particularly when abrupt) commonly leads to withdrawal symptoms.
Dizziness, sensory disturbances (including paraesthesia or electric shock-like sensations, particularly in the head), sleep disturbances (including insomnia and intense dreams), fatigue, somnolence, agitation or anxiety, nausea and/or vomiting, tremor, headache, myalgia, irritability, diarrhoea, hyperhydrosis and vertigo are the most commonly reported reactions.
Generally, for SSRIs and SNRIs, these events are mild to moderate and self-limiting, however, in some patients they may be severe and/or prolonged. It is therefore advised that when duloxetine treatment is no longer required, gradual discontinuation by dose tapering should be carried out (See Dosage & Administration and Precautions).
In the 12 week acute phase of three clinical trials of duloxetine in patients with diabetic neuropathic pain, small but statistically significant increases in fasting blood glucose were observed in duloxetine-treated patients. HbA
1c was stable in both duloxetine-treated and placebo-treated patients. In the extension phase of these studies, which lasted up to 52 weeks, there was an increase in HbA
1c in both the duloxetine and routine care groups, but the mean increase was 0.3% greater in the duloxetine-treated group. There was also a small increase in fasting blood glucose and in total cholesterol in duloxetine-treated patients while those laboratory tests showed a slight decrease in the routine care group.
The heart rate-corrected QT interval in duloxetine-treated patients did not differ from that seen in placebo-treated patients. No clinically significant differences were observed for QT, PR, QRS, or QTcB measurements between duloxetine-treated and placebo-treated patients.
Paediatric population: A total of 509 paediatric patients aged 7 to 17 years with MDD were treated with duloxetine in clinical trials. In general, the adverse reaction profile of duloxetine in children and adolescents was similar to that seen for adults.
A total of 467 paediatric patients initially randomized to duloxetine in clinical trials experienced a 0.1 kg mean decrease in weight at 10-weeks compared with a 0.9 kg mean increase in 353 placebo-treated patients. Subsequently, over the four-to six-month extension period, patients on average trended toward recovery to their expected baseline weight percentile based on population data from age- and gender- matched peers.
In studies of up to 9 months an overall mean decrease of 1% in height percentile (decrease of 2% in children (7-11 years) and increase of 0.3% in adolescents (12-17 years)) was observed in duloxetine-treated paediatric patients (see Precautions).
Monoamine Oxidase Inhibitors (MAOIs): Due to the risk of serotonin syndrome, Cymbalta should not be used in combination with non-selective irreversible monoamine oxidase inhibitors (MAOIs), or within at least 14 days of discontinuing treatment with an MAOI. Based on the half-life of duloxetine, at least 5 days should be allowed after stopping Cymbalta before starting an MAOI (see Contraindications).
The concomitant use of Cymbalta with selective, reversible MAOIs, like moclobemide, is not recommended (see Precautions). The antibiotic linezolid is a reversible non-selective MAOI and should not be given to patients treated with Cymbalta (see Precautions).
Inhibitors of CYP1A2: Because CYP1A2 is involved in duloxetine metabolism, concomitant use of duloxetine with potent inhibitors of CYP1A2 is likely to result in higher concentrations of duloxetine. Fluvoxamine (100 mg once daily), a potent inhibitor of CYP1A2, decreased the apparent plasma clearance of duloxetine by about 77% and increased AUCo-t 6-fold. Therefore Cymbalta should not be administered in combination with potent inhibitors of CYP1A2 like fluvoxamine (see Contraindications).
CNS medicinal products: The risk of using duloxetine in combination with other CNS-active medicinal products has not been systematically evaluated, except in the cases described in this section. Consequently, caution is advised when Cymbalta is taken in combination with other centrally-acting medicinal products or substances, including alcohol and sedative medicinal products (e.g. benzodiazepines, morphinomimetics, antipsychotics, phenobarbital, sedative antihistamines).
Serotonergic agents: In rare cases, serotonin syndrome has been reported in patients using SSRIs/SNRIs concomitantly with serotonergic agents. Caution is advisable if Cymbalta is used concomitantly with serotonergic agents like SSRIs, SNRIs, tricyclic antidepressants like clomipramine or amitriptyline, MAOIs like moclobemide or linezolid, St John’s wort (Hypericum perforatum) or triptans, tramadol, pethidine and tryptophan (see Precautions).
Effect of duloxetine on other drugs: Medicinal products metabolised by CYP1A2: The pharmacokinetics of theophylline, a CYP1A2 substrate,were not significantly affected by co-administration with duloxetine (60 mg twice daily).
Medicinal products metabolised by CYP2D6: Duloxetine is a moderate inhibitor of CYP2D6. When duloxetine was administered at a dose of 60 mg twice daily with a single dose of desipramine, a CYP2D6 substrate, the AUC of desipramine increased 3-fold. The co-administration of duloxetine (40 mg twice daily) increases steady state AUC of tolterodine (2 mg twice daily) by 71 %, but does not affect the pharmacokinetics of its active 5-hydroxyl metabolite and no dosage adjustment is recommended. Caution is advised if Cymbalta is co-administered with medicinal products that are predominantly metabolised by CYP2D6 (risperidone, tricyclic antidepressants [TCAs] such as nortriptyline, amitriptyline, and imipramine) particularly if they have a narrow therapeutic index (such as flecainide, propafenone and metoprolol).
Oral contraceptives and other steroidal agents: Results of in vitro studies demonstrate that duloxetine does not induce the catalytic activity of CYP3A. Specific in vivo drug interaction studies have not been performed.
Anticoagulants and antiplatelet agents: Caution should be exercised when duloxetine is combined with oral anticoagulants or antiplatelet agents due to a potential increased risk of bleeding attributable to a pharmacodynamic interaction. Furthermore, increases in INR values have been reported when duloxetine was co-administered to patients treated with warfarin. However, concomitant administration of duloxetine with warfarin under steady state conditions, in healthy volunteers, as part of a clinical pharmacology study, did not result in a clinically significant change in INR from baseline or in the pharmacokinetics of R- or S-warfarin.
Effects of other drugs on duloxetine: Antacids and H2 antagonists: Co-administration of duloxetine with aluminium- and magnesium- containing antacids or duloxetine with famotidine had no significant effect on the rate or extent of duloxetine absorption after administration of a 40 mg oral dose.
Inducers of CYP1A2: Population pharmacokinetic studies have shown that smokers have almost 50% lower plasma concentrations of duloxetine compared with non-smokers.
Special precautions for disposal and other handling: No special requirements.
Incompatibilities: Not applicable.
Special precautions for storage: Store in the original package in order to protect from moisture. Refer to the recommended storage conditions on the product labels.
N06AX21 - duloxetine ; Belongs to the class of other antidepressants.
Cymbalta gastro-resistant cap 30 mg
28's
Cymbalta gastro-resistant cap 60 mg
28's


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image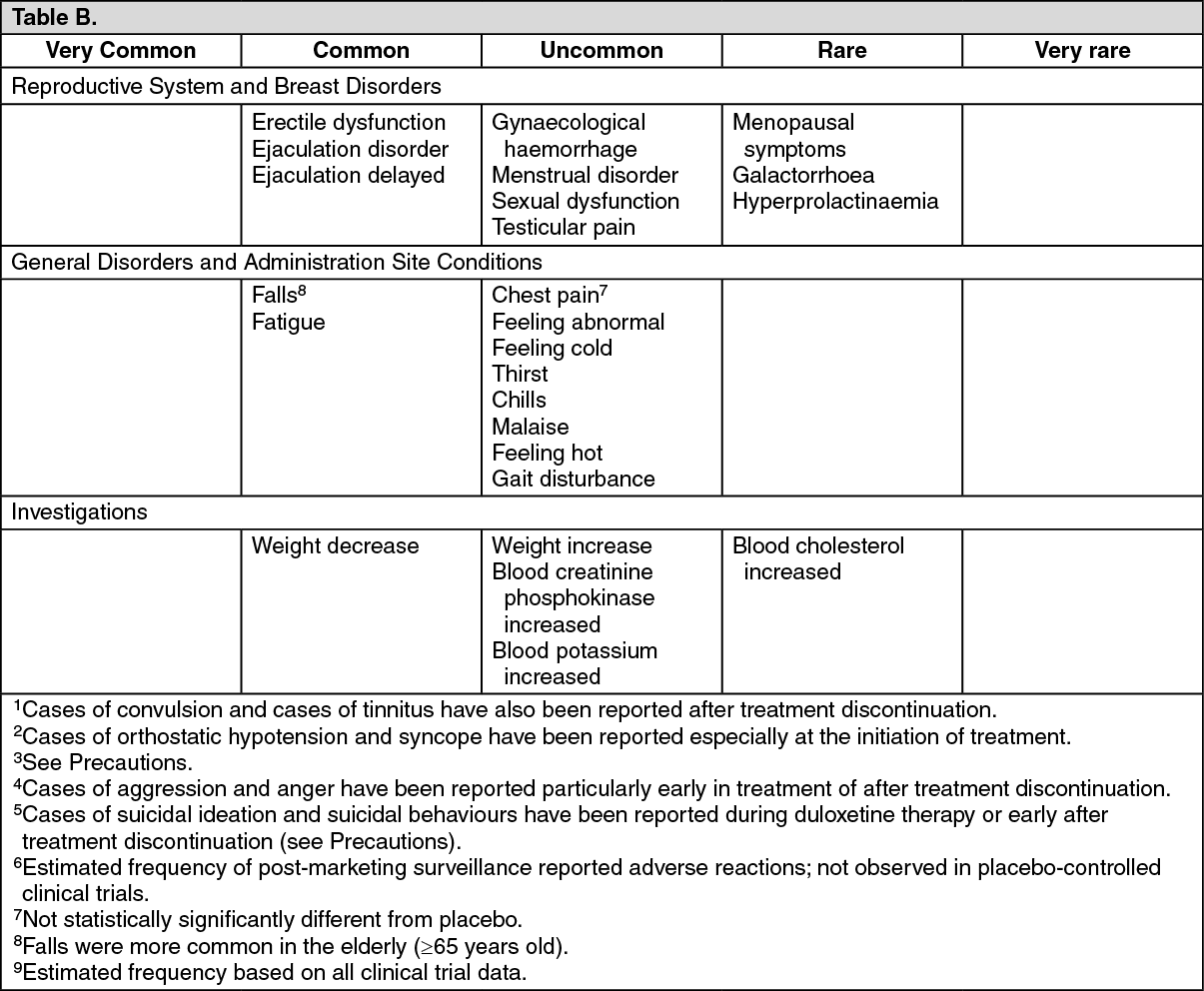 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
