Dosage: Adults: Non small-cell lung cancer:
Single agent use: The recommended dose is 1000 mg/m
2, given by intravenous infusion.
The administration must be repeated once weekly for three weeks, followed by a one-week rest period. This four-week cycle is then repeated. A dose reduction or delay before each administration of the chemotherapy may be applied, based upon the amount of toxicity experienced by the patient.
Combination use: Gemcitabine in combination with cisplatin can be administered using two dosage regimens one regimen use a three-week schedule, the other uses a four-week schedule. The three-week schedule is the usual regimen; this three-week cycle uses gemcitabine 1250 mg/m
2 given by 30 minutes intravenous infusion on days 1 and 8, followed by one-week rest period. This threeweek cycle is then repeated. A dosage reduction or delay before each administration of the chemotherapy may be applied, based upon the amount of toxicity experienced by the patient.
The four-week cycle uses Gemcitabine 1000 mg/m
2 given by 30 minutes intravenous infusion on days 1, 8 and 15, followed by one-week rest period. This four-week cycle is then repeated. A dosage reduction or delay before each administration of the chemotherapy may be applied, based upon the amount of toxicity experienced by the patient.
Pancreatic adenocarcinoma: The recommended dose is 1000 mg/m
2, given by 30 minutes intravenous infusion. This should be repeated once weekly for 7 weeks, followed by a week rest. Subsequent cycles should consist of injections once weekly for 3 consecutive weeks out of every 4 weeks. A dosage reduction or delay before each administration of the chemotherapy may be applied, based upon the amount of toxicity experienced by the patient.
Bladder cancer, at the invasive stage: The recommended dose of Gemcitabine in combination with cisplatin, is 1000 mg/m
2, given by 30 minutes intravenous infusion on days 1, 8 and 15, followed by one-week rest period for a 28 day cycle. Cisplatin is given at a recommended dose of 70 mg/m
2 on day 2. This four-week cycle is then repeated. A dosage reduction or delay before each administration of the chemotherapy may be applied, based upon the amount of toxicity experienced by the patient. A clinical trial showed more myelosuppression when cisplatin was used in doses of 100mg/m
2.
Breast cancer: Paclitaxel (175 mg/m
2) administered on Day 1 over approximately 3 hours as an intravenous infusion, followed by Gemcitabine (1250 mg/m
2) as a 30-minute intravenous infusion on Days 1 and 8 of each 21 day cycle. Dose reduction with each cycle or within a cycle may be applied based upon the amount of toxicity experienced by the patient.
Ovarian Cancer: Gemcitabine in combination with carboplatin is recommended using Gemcitabine 1000 mg/m
2 administered on Days 1 and 8 of each 21-day cycle as a 30-minute intravenous infusion. After Gemcitabine, carboplatin should be given on Day 1 to attain a target AUC of 4.0 mg/mL/min. Dosage reduction with each cycle or within a cycle may be applied based upon the amount of toxicity experienced by the patient.
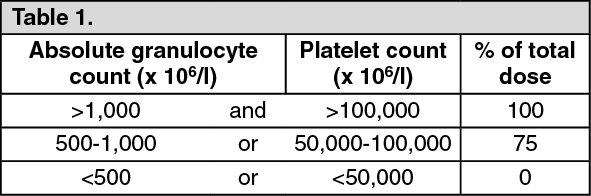
For all indications: Patients receiving Gemcitabine should be monitored prior to each dose for platelet, leukocyte and granulocyte counts and, if necessary, the dose of Gemcitabine may be either reduced or withheld in the presence of hematologic toxicity, according to the following scale: See Table 1.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Periodic physical examination and checks of renal and hepatic function should be made to detect non-hematologic toxicity. Dosage reduction with each cycle or within a cycle may be applied based upon the amount of toxicity experienced by the patient. Doses should be withheld until toxicity has resolved in the opinion of the physician.
Elderly patients: Gemcitabine has been well tolerated by patients over 65 years of age. The pharmacokinetic data suggest that the metabolism of the drug is not affected by age.
Patients with hepatic or renal impairment: Gemcitabine should be used with caution in patients with hepatic insufficiency or with impaired renal function as there is insufficient information from clinical studies to allow clear dose recommendation for this patient population. Mild to moderate renal insufficiency (GFR from 30mL/min to 80mL/min) has no consistent, significant effect on Gemcitabine pharmacokinetics.
Children: Gemcitabine has been studied in limited Phase I and II trials in children in a variety of tumor types. These studies did not provide sufficient data to establish the efficacy and safety of Gemcitabine in children.
Method of Administration: Intravenous route: Gemcitabine is well tolerated during infusion and is usually easy to administer. Reactions at the site of injection are rare: no case of cutaneous necrosis has been reported. If extravasation, the administration must be stopped immediately.
Handling: It is compulsory that injectable solutions of cytotoxic agents be prepared by specialised, trained staff with knowledge of the drugs used, under conditions which ensure protection of the environment, and particularly of the drug handling staff. Preparation requires a room reserved for this purpose. Smoking, eating and drinking are prohibited in this room. The handling staff must have a set of appropriate equipment for handling, particularly long-sleeved coats, protective masks, caps, protective goggles, sterile disposable gloves, worktop protection sheets and waste collection containers and bags. Excreta and vomitus must be handled with care. Pregnant women must be warned and avoid handling cyto-toxic agents. All broken containers must be treated with the same precautions and regarded as contaminated waste. Contaminated waste is to be disposed of by incineration in rigid containers labelled for this purpose.
Instructions for use and handling: The only approved diluent for reconstitution of Gemcitabine sterile powder is 0.9% Sodium Chloride Injection without preservatives. Although no incompatibility has been demonstrated, it is none-the-less recommended that mixing gemcitabine solutions with those of other drugs should be avoided. Due to solubility considerations, the maximum concentration for Gemcitabine upon reconstitution is 40 mg/mL. Reconstitution at concentrations greater than 40 mg/mL may result in incomplete dissolution, and should be avoided.
To reconstitute, add at least 5 ml of 0.9% Sodium Chloride Injection to the 200 mg vial and at least 25 ml of 0.9% Sodium Chloride Injection to the 1000 mg vial. Shake to dissolve. The appropriate amount of drug may be administered as prepared or further diluted with 0.9% Sodium Chloride Injection. Parenteral drugs should be inspected visually for particulate matter and discoloration, prior to administration. As other cystostatics, Gemcitabine hydrochloride must be handled with care. Unused products must be destroyed according to hospital procedures of cytotoxic waste deal.
Route of Administration: Parenteral.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image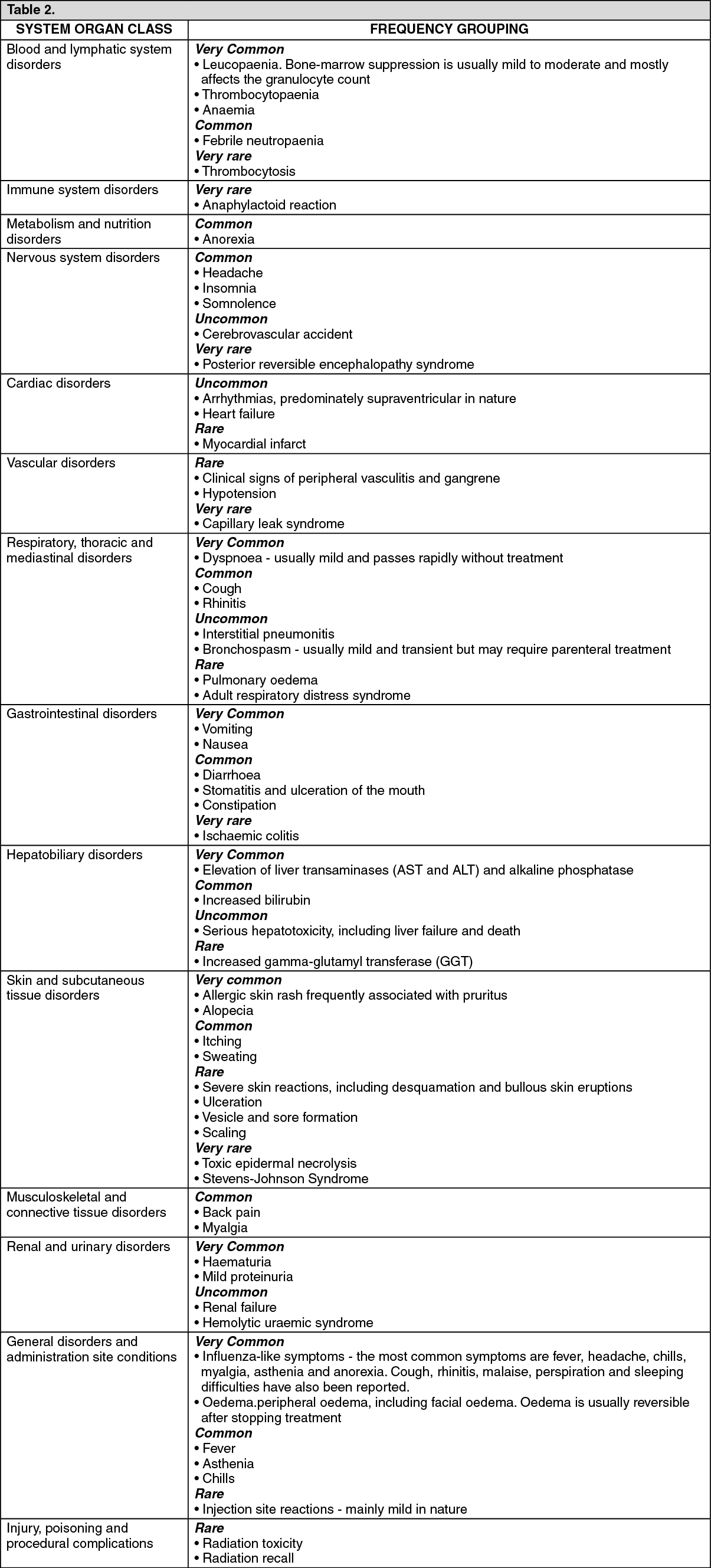 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
