Summary of the safety profile: Adult population with MDS, CMML and AML (20-30% marrow blasts): Adverse reactions considered to be possibly or probably related to the administration of Vidaza have occurred in 97 % of patients.
The most common serious adverse reactions included febrile neutropenia and anaemia. Other serious adverse reactions included infections such as neutropenic sepsis and pneumonia (some with fatal outcome), thrombocytopenia, hypersensitivity reactions and haemorrhagic events (e.g. cerebral haemorrhage, gastrointestinal haemorrhage and intracranial haemorrhage.
The most commonly reported adverse reactions with azacitidine treatment were haematological reactions including thrombocytopenia, neutropenia and leukopenia (usually Grade 3-4), gastrointestinal events including nausea, vomiting (usually Grade 1-2) or injection site reactions (usually Grade 1-2).
Adult population aged 65 years or older with AML with > 30% marrow blasts: The most common serious adverse reactions in the azacitidine treatment arm included febrile neutropenia, pneumonia, and pyrexia. Other less frequently reported serious adverse reactions in the azacitidine treatment arm included sepsis, anemia, neutropenic sepsis, urinary tract infection, thrombocytopenia, neutropenia, cellulitis, dizziness and dyspnoea.
The most commonly reported adverse reactions with azacitidine treatment were gastrointestinal events, including constipation, nausea, and diarrhoea, (usually Grade 1-2), general disorders and administration site conditions including pyrexia (usually Grade 1-2) and haematological events, including febrile neutropenia and neutropenia, (usually Grade 3-4).
Tabulated list of adverse reactions: Table 3 as follows contains adverse reactions associated with azacitidine treatment obtained from the main clinical studies in MDS and AML and post marketing surveillance.
Adverse reactions are presented in the table as follows according to the highest frequency observed in any of the main clinical studies. (See Table 3.)
 Click on icon to see table/diagram/image
Description of selected adverse reactions: Haematologic adverse reactions:
Click on icon to see table/diagram/image
Description of selected adverse reactions: Haematologic adverse reactions: The most commonly reported haematological adverse reactions associated with azacitidine treatment include anaemia, thrombocytopenia, neutropenia, febrile neutropenia and leukopenia, and were usually Grade 3 or 4. There is a greater risk of these events occurring during the first 2 cycles, after which they occur with less frequency in patients with restoration of haematological function. Most haematological adverse reactions were managed by routine monitoring of complete blood counts and delaying azacitidine administration in the next cycle, prophylactic antibiotics and/or growth factor support (e.g. G-CSF) for neutropenia and transfusions for anaemia or thrombocytopenia as required.
Infections: Myelosuppression may lead to neutropenia and an increased risk of infection. Serious adverse reactions such as sepsis, including neutropenic sepsis, and pneumonia were reported in patients receiving azacitidine, some with a fatal outcome. Infections may be managed with the use of anti-infectives plus growth factor support (e.g. G-CSF) for neutropenia.
Bleeding: Bleeding may occur with patients receiving azacitidine. Serious adverse reactions such as gastrointestinal haemorrhage and intracranial haemorrhage have been reported. Patients should be monitored for signs and symptoms of bleeding, particularly those with pre-existing or treatment-related thrombocytopenia.
Hypersensitivity: Serious hypersensitivity reactions have been reported in patients receiving azacitidine. In case of an anaphylactic-like reaction, treatment with azacitidine should be immediately discontinued and appropriate symptomatic treatment initiated.
Skin and subcutaneous tissue adverse reactions: The majority of skin and subcutaneous adverse reactions were associated with the injection site. None of these adverse reactions led to discontinuation of azacitidine, or reduction of azacitidine dose in the pivotal studies. The majority of adverse reactions occurred during the first 2 cycles and tended to decrease with subsequent cycles. Subcutaneous adverse reactions such as injection site rash/inflammation/pruritus, rash, erythema and skin lesion may require management with concomitant medicinal products, such as antihistamines, corticosteroids and non-steroidal anti-inflammatory medicinal products (NSAIDs). These cutaneous reactions have to be distinguished from soft tissue infections, sometimes occurring at injection site. Soft tissue infections, including cellulitis and necrotising fasciitis in rare cases leading to death, have been reported with azacitidine in the post marketing setting.
Gastrointestinal adverse reactions: The most commonly reported gastrointestinal adverse reactions associated with azacitidine treatment included constipation, diarrhoea, nausea and vomiting. These adverse reactions were managed symptomatically with anti-emetics for nausea and vomiting; anti-diarrhoeals for diarrhoea, and laxatives and/or stool softeners for constipation.
Renal adverse reactions: Renal abnormalities, ranging from elevated serum creatinine and haematuria to renal tubular acidosis, renal failure and death were reported in patients treated with azacitidine.
Hepatic adverse reactions: Patients with extensive tumour burden due to metastatic disease have been reported to experience hepatic failure, progressive hepatic coma and death during azacitidine treatment.
Cardiac events: Patients with known history of cardiovascular or pulmonary disease showed a statistically significant increase in cardiac events in patients with newly diagnosed AML treated with azacitidine.
Elderly population: There is limited safety information available with azacitidine in patients ≥85 years.


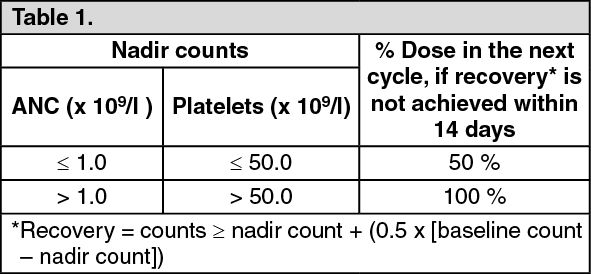 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image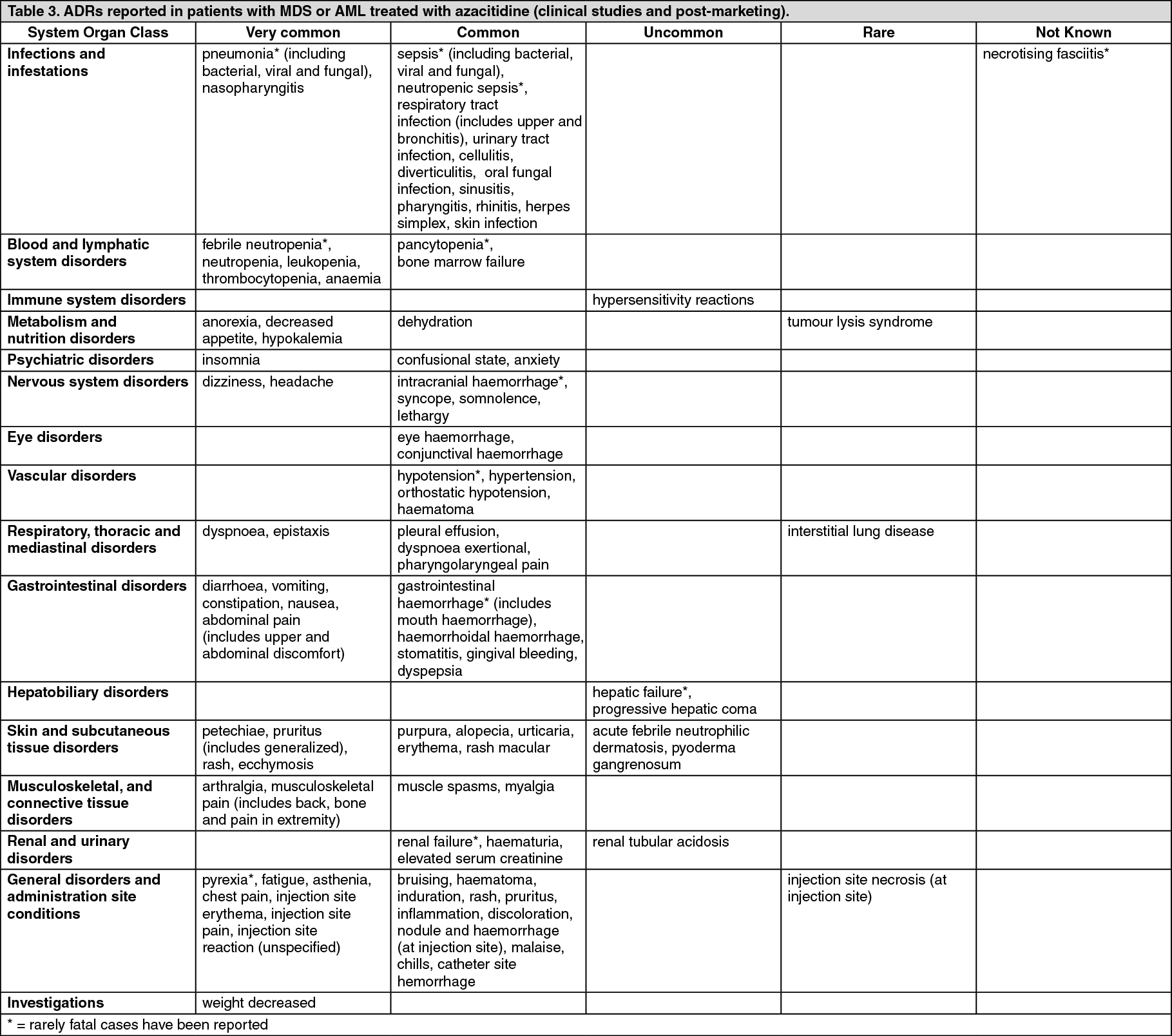 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
