Pharmacology: Mechanism of Action: ADYNOVATE, a PEGylated form of recombinant antihemophilic factor (ADVATE), [see Description], temporarily replaces the missing coagulation factor VIII needed for effective hemostasis in congenital hemophilia A patients. ADYNOVATE exhibits an extended terminal half-life through pegylation of the parent molecule, ADVATE, which reduces binding to the physiological factor VIII clearance receptor (LRP1).
Pharmacodynamics: Hemophilia A is a disorder characterized by a deficiency of functional coagulation factor VIII, resulting in a prolonged, patient plasma clotting time as measured by the activated partial thromboplastin time (aPTT). Treatment with ADYNOVATE normalizes the aPTT over the effective dosing period. The administration of ADYNOVATE increases plasma levels of factor VIII and can temporarily correct the coagulation defect in hemophilia A patients.
CLINICAL STUDIES: Original Safety and Efficacy Clinical Trial: The safety, efficacy, and PK of ADYNOVATE were evaluated in a multicenter, open-label, prospective, non-randomized, two-arm clinical trial that compared the efficacy of a twice weekly prophylactic treatment regimen to on-demand treatment and determined hemostatic efficacy in the treatment of bleeding episodes. A total of 137 male PTPs (12 to 65 years of age) with severe hemophilia A received at least one infusion with ADYNOVATE. Twenty-five of the 137 subjects were adolescents (12 to less than 18 years of age).
Subjects received either prophylactic treatment (n = 120) with ADYNOVATE at a dose of 40-50 IU per kg twice weekly or on-demand treatment (n = 17) with ADYNOVATE at a dose of 10-60 IU per kg for a 6-month period. The mean (SD) dose per prophylaxis infusion was 44.4 (3.9) IU per kg with a median dosing interval of 3.6 days. There were 91 out of 98 (93%) subjects previously treated prophylactically prior to enrollment, who experienced a reduction in dosing frequency during routine prophylaxis in the trial, with a median reduction of 33.7% (approximately one more day between doses). One hundred eighteen of 120 (98%) prophylaxis subjects remained on the starting recommended regimen without dose adjustment, and 2 subjects increased their dose to 60 IU/kg during prophylaxis due to bleeding in target joints.
On-demand Treatment and Control of Bleeding Episodes: A total of 518 bleeding episodes were treated with ADYNOVATE in the per-protocol population, i.e. dosed according to the protocol specific dosing requirements. Of these, 361 bleeding episodes (n=17 subjects) occurred in the on-demand arm and 157 (n=61 subjects) occurred in the prophylaxis arm. The median dose per infusion to treat all bleeding episodes in the per-protocol population was 29 (Q1: 20.0; Q3: 39.2) IU per kg. The median dose per infusion to treat a minor, moderate, or severe/major bleeding episode in the per-protocol population was 25.5 (Q1: 16.9; Q3: 37.6) IU/kg, 30.9 (Q1: 23.0; Q3: 43.1) IU/kg, or 36.4 (Q1: 29.0; Q3: 44.5) IU/kg, respectively.
A total of 591 bleeding episodes were treated with ADYNOVATE in the treated population, which was identical to the safety analysis set of subjects assigned to routine prophylaxis or on-demand treatment with ADYNOVATE and who received at least one dose of the product. Of these, 361 bleeding episodes (n=17 subjects) occurred in the on-demand arm and 230 bleeding episodes (n=75 subjects) occurred in the routine prophylaxis arm. Efficacy in control of bleeding episodes is summarized in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Routine Prophylaxis: A total of 120 subjects (treated population) received a twice a week regimen in the prophylaxis arm, and an additional 17 subjects were treated episodically in the on-demand arm. In the treated population, the median [mean] annualized bleed rate (ABR) in the on-demand treatment arm was 41.5 [40.8] compared to 1.9 [4.7] while on a twice a week prophylaxis regimen (Table 3). In the per-protocol population, the median [mean] annualized bleed rate (ABR) in the on-demand treatment arm was 41.5 [40.8] compared to 1.9 [3.7] while on a twice a week prophylaxis regimen. Using a negative binomial model to estimate the ABR, there was a significant reduction in the ABR (p <0.0001) for subjects in the prophylaxis arm compared to the on-demand arm. (See Table 3.)
Click on icon to see table/diagram/image
In the treated population, the median [mean] ABR for the 23 adolescent subjects age 12 to <18 years of age on routine prophylaxis was 2.1 [5.2] compared to a median [mean] ABR of 1.9 [4.6] for the 97 subjects 18 years and older. Reduction in ABR between the treatment arms was observed regardless of baseline subgroups examined, including age, presence or absence of target joints, and pre-trial treatment regimen. The majority of the bleeding episodes during prophylaxis (95%) were of minor/moderate severity. Forty-five out of 120 subjects (38%) experienced no bleeding episodes and 68 out of 120 subjects (57%) experienced no joint bleeding episodes in the prophylaxis arm. Of those subjects who were compliant to regimen (per-protocol population), 40 out of 101 subjects (40%) experienced no bleeding episodes. All subjects in the on-demand arm experienced a bleeding episode, including a joint bleeding episode.
Routine Prophylaxis Clinical Trial in Pediatric Subjects (<12 years of age): The safety and efficacy of ADYNOVATE was evaluated in a total of 73 pediatric PTPs with severe hemophilia A, of which 66 subjects were dosed (32 subjects aged <6 years and 34 subjects aged 6 to <12 years) in a separate pediatric clinical trial. The prophylactic regimen was 40 to 60 IU/kg of ADYNOVATE twice a week, with a mean (SD) dose of 51.1 IU/kg (5.5). The median [mean] overall ABR was 2.0 [3.61] for the 66 subjects in the treated population and the median [mean] ABRs for spontaneous and joint bleeding episodes were both 0 [1.18 and 1.12, respectively]. Of the 66 subjects treated prophylactically, 25 (38%) experienced no bleeding episodes, 44 (67%) experienced no spontaneous bleeding episodes, and 48 (73%) experienced no joint bleeding episodes.
Of the 70 bleeding episodes observed during the pediatric trial, 82.9% were controlled with 1 infusion and 91.4% were controlled with 1 or 2 infusions. Control of bleeding was rated excellent or good in 63 out of 70 (90%) bleeding episodes. The definitions of excellent or good in the pediatric clinical trial were unchanged as compared to the previously conducted prophylaxis clinical trial in adolescent and adult subjects.
An extension study in adult and pediatric patients evaluated the safety and efficacy of prophylactic treatment regimen in 216 previously treated patients with severe hemophilia A. Majority had completed the adult and adolescent study or the pediatric study. Similar efficacy was noted in this extension study.
Perioperative Management Clinical Trial: Twenty-one major surgical procedures comprised of 14 orthopedic, and 7 non-orthopedic procedures, and 5 additional minor surgeries were performed in 21 subjects. The preoperative loading dose ranged from 36 IU/kg to 99 IU/kg (median: 60 IU/kg) and the total postoperative dose ranged from 23 IU/kg to 769 IU/kg (median: 183 IU/kg). The median total dose (including all administrations from pre-surgical PK and loading doses to post-hospital follow up) was 629 IU/kg (range: 464 - 1,457 IU/kg) for major orthopedic surgeries, 489 IU/kg (range: 296 - 738 IU/kg) for major non-orthopedic surgeries.
Overall hemostatic efficacy was rated as excellent [blood loss less than or equal to that expected for the same type of procedure performed in a non-hemophilic patient, for all 24 (21 major, 3 minor)] procedures with available assessments.
Pharmacokinetics: The pharmacokinetics (PK) of ADYNOVATE were evaluated in a multi-center, prospective, open label clinical trial and compared with ADVATE in 26 subjects prior to initiation of prophylactic treatment with ADYNOVATE and in 22 subjects after 6 months of treatment with ADYNOVATE. A single dose of 45 IU/kg was utilized for both products. The PK parameters, as shown in Table 4, were based on plasma coagulation factor VIII activity measured by the one-stage clotting assay and are presented by age groups.
Incremental recovery was comparable between both products. The PK parameters determined after 6 months of prophylactic treatment with ADYNOVATE were consistent with the initial parameter estimates.
Pediatric Pharmacokinetics: Pharmacokinetic parameters calculated from 39 subjects <18 years of age (intent-to-treat analysis) are available for 14 children (2 to <6 years), 17 older children (6 to <12 years) and 8 adolescent subjects (12 to <18 years of age), as shown in Table 4. The mean clearance (based on body weight) of ADYNOVATE was higher and the mean half-life was lower in children <12 years of age than adults. A dose adjustment may be required in children <12 years of age. (See Table 4.)
Click on icon to see table/diagram/image
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Long-term studies in animals to evaluate the carcinogenic potential of ADYNOVATE or studies to determine the effects of ADYNOVATE on genotoxicity or fertility have not been performed.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image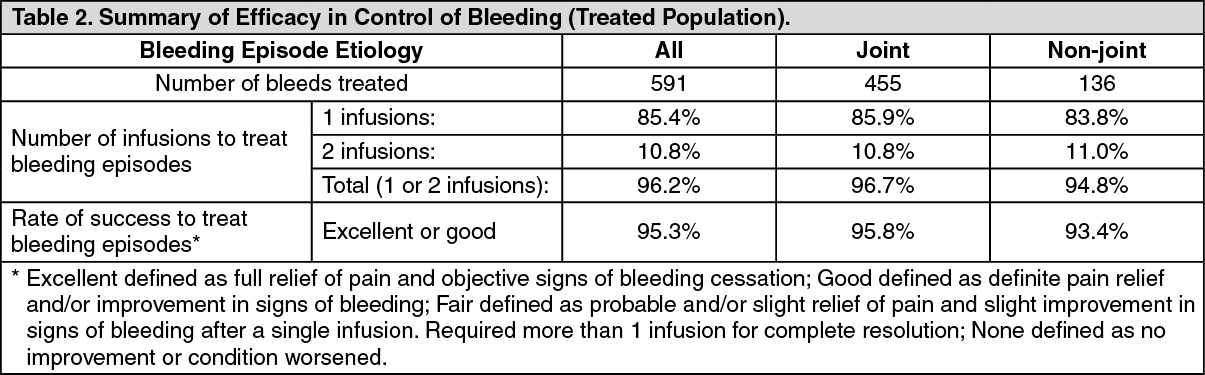 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image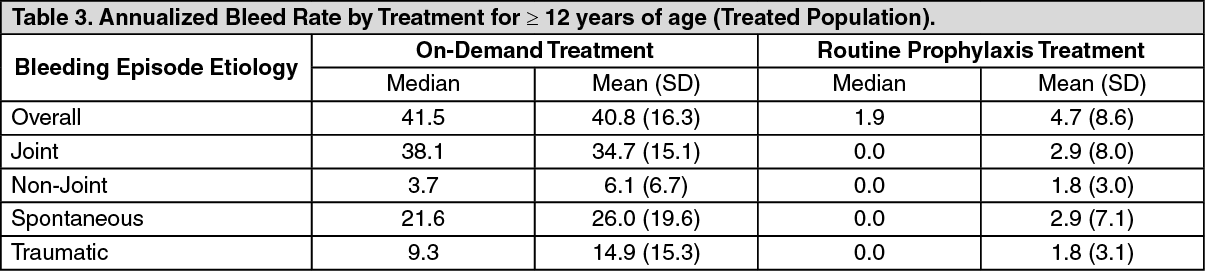 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image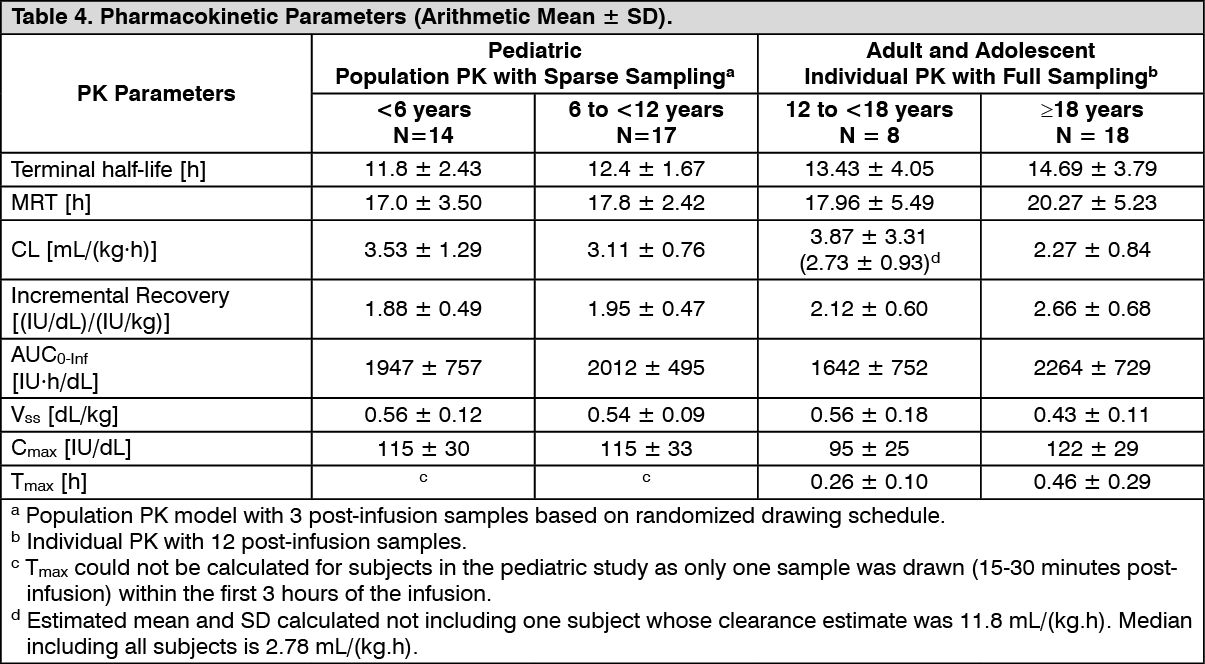 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image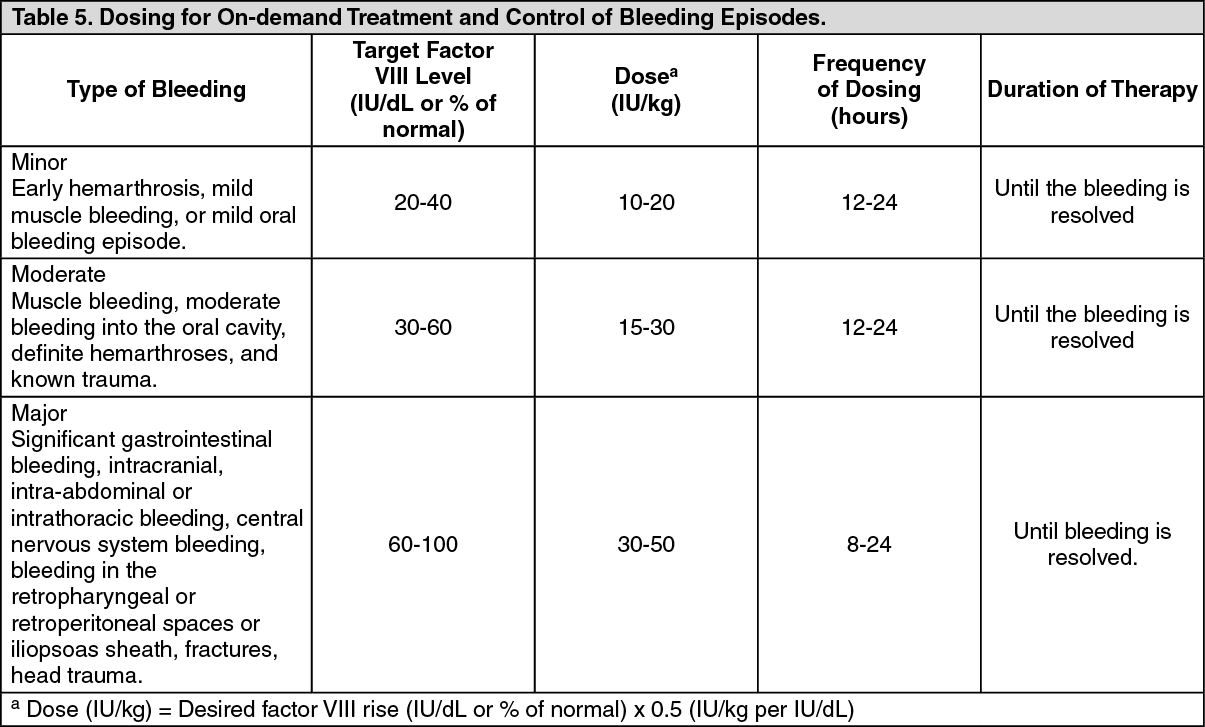 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image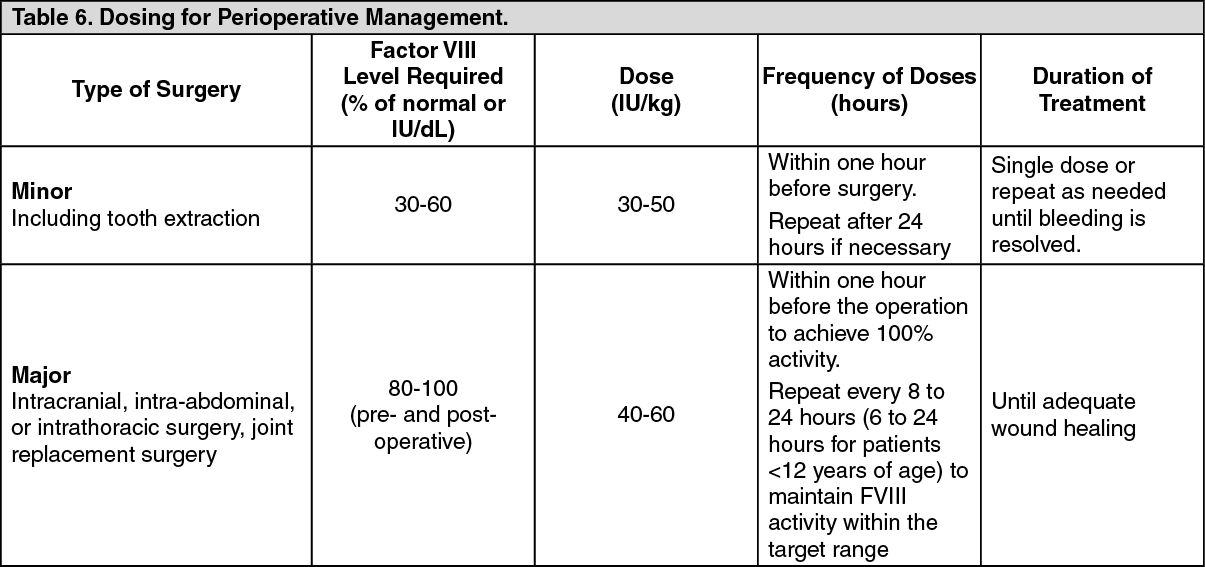 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image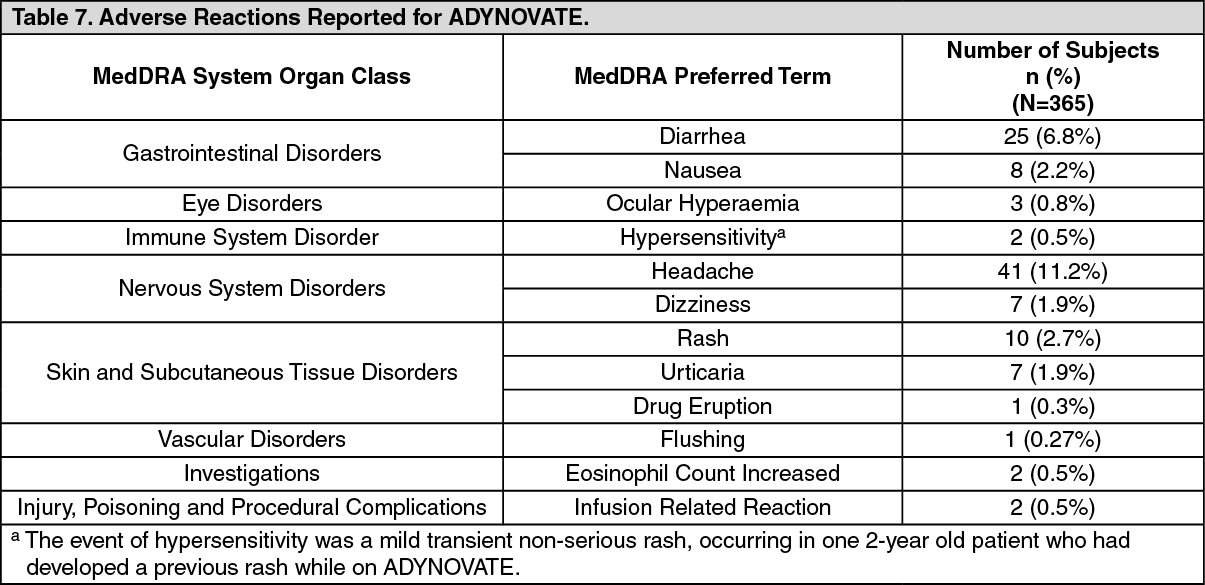 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
