Pharmacologic class: CD30-directed antibody-drug conjugate.
Pharmacotherapeutic group: monoclonal antibodies.
ATC code: L01XC12.
Pharmacology: Mechanism of Action: Adcetris is an Antibody Drug Conjugate (ADC) that delivers an antineoplastic agent that result in apoptotic cell death selectively in CD30-expressing tumor cells. Nonclinical data suggest that the biological activity of Adcetris results from a multi-step process. Binding of the ADC to CD30 on the cell surface initiates internalization of the ADC-CD30 complex, which then trafficks to the lysosomal compartment. Within the cell, a single defined active species, MMAE, is released via proteolytic cleavage. Binding of MMAE to tubulin disrupts the microtubule network within the cell, induces cell cycle arrest and results in apoptotic death of the CD30-expressing tumor cell.
Contributions to the mechanism of action by other antibody associated functions have not been excluded.
Pharmacodynamics: Pharmacodynamics Effects (e.g. subsections: Resistance, In vitro Susceptibility Data): General: No primary pharmacodynamic relationships have been identified.
Cardiac Electrophysiology:
Forty-six (46) patients with CD30-expressing hematologic malignancies were evaluable of the 52 patients who received 1.8 mg/kg of Adcetris every 3 weeks as part of a phase 1, single-arm, open-label, multicenter cardiac safety study. The primary objective was to evaluate the effect of Adcetris on cardiac ventricular repolarization and the predefined primary analysis was the change in QTc from baseline to multiple time points in Cycle 1.
The upper 90% confidence interval (CI) was <10 msec at each of the Cycle 1 post-baseline time-points. These data indicate the absence of clinically relevant QT prolongation due to Adcetris administered at a dose of 1.8 mg/kg in patients with CD30-expressing malignancies.
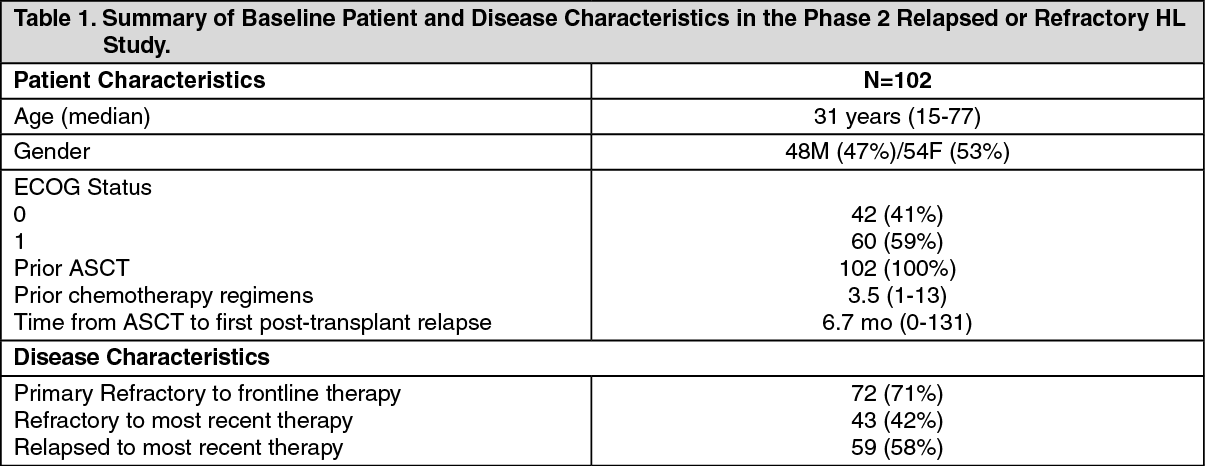
CLINICAL STUDIES: Hodgkin Lymphoma: Study SG035-0003: The efficacy and safety of Adcetris as a single agent was evaluated in an open-label, single-arm, multicenter study in 102 patients with relapsed or refractory Hodgkin Lymphoma (HL). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
All patients had a histologically confirmed CD30-expressing disease and had at least one prior autologous stem cell transplant (ASCT). Seventy-two patients (71%) had primary refractory HL, defined as a failure to achieve a complete response to, or progressed within 3 months of completing frontline therapy; 43 patients (42%) were refractory and 59 patients (58%) had relapsed following their most recent prior therapy. Patients had received a median of 3.5 prior systemic chemotherapies. The median time from ASCT to first post-transplant relapse was 6.7 months. Patients received up to 16 cycles of therapy; the median number of cycles received was 9 (ranging from 1 to 16). The primary endpoint, Objective Response Rate, was 74.5%. See Table 2 as follows for other pre-specified endpoints. (See Table 2.)
Click on icon to see table/diagram/image
No clinically meaningful differences in the objective response rate were observed within the subgroups analyzed among the following subgroups analyzed: gender, baseline weight (≤100 kg versus >100 kg), baseline B symptoms, number of treatments prior to ASCT (≤2 versus >2), number of treatments post-ASCT (0 versus ≥1), relapsed versus refractory to last therapy, primary refractory disease, and time from ASCT to relapse post-ASCT (≤1 year versus >1 year).
Tumor reduction was achieved in 94% of patients. See Figure 1 for waterfall chart of tumor reduction, ORR and CR. (See Figure 1.)
Click on icon to see table/diagram/image
Per IRF, median time to first response was 1.3 months, and median time to CR was 2.8 months. Median duration of objective response was 6.7 months (95% CI [3.6, 14.8]) with a range of 1.2+ to 26.1+ months. Of the patients treated, 7 responding patients went on to receive an allogeneic stem cell transplant.
Of the 35 patients who had B symptoms at baseline, 27 patients (77%) experienced resolution of all B symptoms at a median time from initiation of Adcetris of 0.7 months.
Per IRF, the median PFS for patients treated with Adcetris was 5.6 months (95% CI [5.0, 9.0]) (the median follow-up time from first dose for patients who were censored on PFS was 5.8 months). Patients who attained a CR achieved a median PFS of 29.2 months while those who attained a PR achieved a median PFS of 5.1 months and those who attained SD achieved a median PFS of 3.5 months. See Figure 2 for median PFS by best clinical response. (See Figure 2.)
Click on icon to see table/diagram/image
Patients who received Adcetris achieved a PFS improvement versus their most recent post ASCT therapy (7.8 months [5.2, 9.9] versus 4.1 months [3.4, 4.9] as assessed by investigator). See Figure 3 for a KM plot of PFS with Adcetris compared to PFS from most recent post-ASCT therapy. (See Figure 3.)
Click on icon to see table/diagram/image
In addition, patients experienced a greater overall and complete response rate compared to their most recent post-ASCT therapy. The median overall survival was 40.5 months.
An exploratory intra-patient analysis showed that approximately 64% of the HL patients treated with brentuximab vedotin as part of the SG035-0003 clinical study experienced an improvement in clinical benefit as measured by longer progression free survival (PFS) compared with their most recent prior line of therapy.
Data were collected from patients (n=15) in phase 1 dose escalation and clinical pharmacology studies, and from patients (n=26) in a Named-Patient Program (NPP), with relapsed or refractory HL who had not received an ASCT, and who were treated with 1.8 mg/kg of Adcetris every 3 weeks.
Baseline patient characteristics showed failure from multiple prior chemotherapy regimens (median of 3 with a range of 1 to 7) before first administration with brentuximab vedotin. Fifty nine percent (59%) of patients had advanced stage disease (stage III or IV) at initial diagnosis.
Results from these phase 1 studies and from the NPP experience showed, that in patients with relapsed or refractory HL without prior ASCT, clinically meaningful responses can be achieved as evidenced by an investigator-assessed, objective response rate of 54% and a complete remission rate of 22% after a median of 5 cycles of brentuximab vedotin.
Study SGN35-005: The efficacy and safety of brentuximab vedotin were evaluated in a randomized, double-blinded, placebo-controlled, 2-arm multicenter trial in 329 patients with HL at risk of relapse or progression following ASCT. Of the 329 patients, 165 patients were randomized to the treatment arm and 164 patients were randomized to the placebo arm. The safety population in the Adcetris arm (N=167) included two additional patients who received at least one dose of Adcetris but were not randomized to the treatment arm. In the study, patients were to receive their first dose after recovery from ASCT (between days 30-45 following ASCT). Patients were treated with 1.8 mg/kg of Adcetris or matching placebo intravenously over 30 minutes every 3 weeks for up to 16 cycles. The median number of cycles received in both arms was 15 cycles. Eligible patients were required to have at least one of the following risk factors: HL that was refractory to frontline treatment; Relapsed or progressive HL that occurred <12 months from the end of frontline treatment; Extranodal involvement at time of pre-ASCT relapse, including extranodal extension of nodal masses into adjacent vital organs. (See Tables 3 and 4, Figures 4 and 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pre-specified subgroup analyses of PFS per IRF were performed by patients' best response to pre-ASCT salvage therapy, HL status after frontline therapy, age, gender, baseline weight, baseline ECOG performance status, number of treatments pre-ASCT, geographic region, pre-ASCT PET status, B symptom status after failure of frontline therapy, and pre-ASCT extranodal disease status. The analyses showed a consistent trend towards benefit for patients who received placebo with the exception of patients ≥ 65 years of age (n=8).
At the time of primary PFS analysis, an interim OS analysis was performed and there was no significant difference in OS between the treatment and placebo arms. Fifty-three patients had died; 28/165 patients in the brentuximab vedotin arm versus 25/164 patients in the placebo arm.
Quality of life was assessed using the EQ-5D instrument. No clinically meaningful differences were observed between the treatment and placebo arms.
Systemic Anaplastic Large Cell Lymphoma (sALCL): Study SG035-0004: The efficacy and safety of Adcetris as a single agent was evaluated in an open-label, single-arm, multicenter study in 58 patients with relapsed or refractory sALCL. (See Table 5.)
Click on icon to see table/diagram/image
All patients had a histologically confirmed CD30-expressing disease and had received front-line chemotherapy with curative intent. A total of 58 patients were treated: 36 patients (62%) had primary refractory sALCL, defined as a failure to achieve a complete response to, or progressed within 3 months of completing frontline therapy; 29 patients (50%) were relapsed and 29 patients (50%) were refractory to most recent prior therapy; 42 patients (72%) had anaplastic lymphoma kinase (ALK)-negative disease. Patients had received a median of 2 prior systemic chemotherapies. Fifteen patients (26%) had received a prior ASCT. The median time from initial sALCL diagnosis to first dose with Adcetris was 16.8 months. Patients received up to 16 cycles of therapy; the median number of cycles received was 7 (range, 1 to 16). The primary endpoint, Objective Response Rate, was 86.2%. See Table 7 as follows for other pre-specified endpoints. (See Table 6.)
Click on icon to see table/diagram/image
No clinically meaningful differences in the objective response rate were observed among the following subgroups analyzed: gender, baseline weight (≤100 kg versus >100 kg), baseline B symptoms, prior autologus stem cell transplantation (ASCT), and post-treatment ASCT. The ORR for relapsed patients was higher than those who were refractory (97% vs. 76%).
Tumor reduction was achieved in 97% of patients. See Figure 4 for waterfall chart of tumor reduction, ORR and CR. (See Figure 6.)
Click on icon to see table/diagram/image
Per IRF, median time to first objective response was 1.4 months (range, 1.0 - 3.2 months) and the median time to CR was 2.7 months (range, 1.2 - 11.6 months). Median duration of objective response was 13.2 months (95% CI [5.7, NE]) with a range of 0.1+ to 21.7+ months (the median follow-up time from first dose was 11.8 months). Of the patients treated, 9 responding patients went on to receive an allogeneic stem cell transplant (SCT) and 7 responding patients went onto autologous SCT.
Of the 17 patients who had B symptoms at baseline, 14 patients (82%) experienced resolution of all B symptoms in a median time from initiation of Adcetris of 0.7 months.
Per IRF, the median PFS for patients treated with Adcetris was 14.6 months (the median follow-up time from first dose was 14.2 months). Patients who attained a CR achieved a median PFS of 27.4 months while those who attained a PR achieved a PFS of 3.9 months. See Figure 7 for median PFS by best clinical response. (See Figure 7.)
Click on icon to see table/diagram/image
Patients who received Adcetris achieved a PFS improvement versus last therapy received prior to study entry (19.6 months [9.1, -NE] versus 5.9 months [3.9, 8.3] as assessed by investigator). See Figure 8 for a KM plot of PFS with Adcetris compared to PFS from last therapy received prior to study entry. (See Figure 8.)
Click on icon to see table/diagram/image
In addition, patients experienced a greater overall and CR rate compared to their most recent therapy. The median overall survival was not reached. The estimated 36 months overall survival was 63% (95% CI [51, 76]).
An exploratory intra-patient analysis showed that approximately 69% of the sALCL patients treated with brentuximab vedotin as part of the SG035-0004 clinical study experienced an improvement in clinical benefit as measured by longer progression free survival (PFS) compared with their most recent prior line of therapy.
Cutaneous T-Cell Lymphoma (CTCL): Study C25001: The efficacy and safety of Adcetris as a single agent was evaluated in a pivotal phase 3, open-label, randomized, multicenter study in 128 patients with histologically confirmed CD30-expressing CTCL.
Patients were stratified by disease subtype (mycosis fungoides [MF] or primary cutaneous anaplastic large cell lymphoma [pcALCL]) and randomized 1:1 to receive either Adcetris or the physician's choice of either methotrexate or bexarotene. Patients with pcALCL received either prior radiation therapy or at least 1 prior systemic therapy and patients with MF received at least 1 prior systemic therapy. Patients were treated with 1.8 mg/kg of Adcetris intravenously over 30 minutes every 3 weeks for up to 16 cycles or physician's choice for up to 48 weeks. The median number of cycles was approximately 12 cycles in the Adcetris arm. In the physician's choice arm, the median duration of treatment (number of cycles) for patients receiving bexarotene was approximately 16 weeks (5.5 cycles) and 11 weeks (3 cycles) for patients receiving methotrexate. Table 7 provides a summary of the baseline patient and disease characteristics. (See Table 7.)
Click on icon to see table/diagram/image
The primary endpoint was objective response rate that lasts at least 4 months (ORR4) (duration from first response to last response ≥ 4 months), as determined by an independent review of the Global Response Score (GRS) consisting of skin evaluations (modified severity weighted assessment tool [mSWAT] assessment), nodal and visceral radiographic assessment, and detection of circulating Sézary cells. The ORR4 was significantly higher in the Adcetris arm compared to the physician's choice arm (56.3% vs 12.5%, p<0.0001). Table 8 includes the results for ORR4 and other key secondary endpoints. (See Table 8.)
Click on icon to see table/diagram/image
Pre-specified subgroup analyses of ORR4 per IRF were performed by patients' CTCL subtype, physicians' choice of treatment, baseline ECOG status, age, gender, and geographic region. The analyses showed a consistent trend towards benefit for patients who received Adcetris compared with patients who received physician's choice. ORR4 was 50% and 75% in the Adcetris arm versus 10.2% and 20% in the physician's choice arm for MF and pcALCL, respectively.
No meaningful differences in quality of life (assessed by the EuroQol five dimensions questionnaire [EQ-5D] and Functional Assessment of Cancer Therapy-General [FACT-G]) were observed between the treatment arms.
The efficacy and safety of Adcetris were evaluated in two additional open-label studies in 108 patients with relapsed CD30+ CTCL (including patients with MF and pcALCL as well as SS, Lyp and mixed CTCL histology) regardless of CD30 expression level. Patients were treated with Adcetris 1.8 mg/kg intravenously over 30 minutes every 3 weeks for up to 16 cycles. The safety and efficacy results in these studies were consistent with results in Study C25001. Overall response rates for MF were 54-66%; pcALCL, 67%; SS, 50%; LyP, 92%; and mixed CTCL histology, 82-85%.
Hodgkin Lymphoma: Study C25003: The efficacy and safety of Adcetris were evaluated in a randomized, open-label, 2-arm, multicenter trial in 1334 patients with advanced frontline HL in combination with chemotherapy (doxorubicin [A], vinblastine [V] and dacarbazine [D] [AVD]). All patients had CD30-expressing HL. Sixty-two percent of patients had extranodal site involvement. Of the 1334 patients, 664 patients were randomized to the Adcetris + AVD arm and 670 patients were randomized to the ABVD (doxorubicin [A], bleomycin [B], vinblastine [V] and dacarbazine [D]) arm and stratified by the number of International Prognostic Factor Project (IPFP) risk factors and region. Patients were treated with 1.2 mg/kg of Adcetris administered as an intravenous infusion over 30 minutes on days 1 and 15 of each 28-day cycle + AVD. The median number of cycles received was 6 (range, 1 to 6 cycles). Table 9 provides a summary of the baseline patient and disease characteristics. (See Table 9.)
Click on icon to see table/diagram/image
The primary endpoint in Study C25003 was modified PFS per IRF, defined as time from randomization to progression, death, or evidence of non-CR after completion of frontline therapy per independent review facility (IRF) followed by subsequent anticancer therapy. Timing of the modified event was the date of the first PET scan post completion of frontline therapy demonstrating the absence of CR, defined as Deauville score of ≥3. The median mPFS by IRF assessment was not estimable for either treatment arm.
The results showed a statistically significant improvement in modified PFS for Adcetris+AVD, with a 2-sided p-value of 0.035 based on a stratified log-rank test. The stratified hazard ratio was 0.770 (95% CI, 0.603; 0.983), indicating a 23% reduction in the risk of modified PFS events for Adcetris+AVD versus ABVD. Table 10 provides the efficacy results for modified PFS and overall survival (OS). (See Table 10, Figures 9 and 10.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Other secondary efficacy endpoints including CR rate and ORR at the end of randomization regimen, CR rate at the end of frontline therapy, and the rate of PET negativity at the end of Cycle 2, duration of response (DOR), duration of complete remission (DOCR), disease-free survival (DFS,) and event-free survival (EFS) all trended in favor of Adcetris+AVD.
Pre-specified subgroup analyses of modified PFS per IRF were performed. The analyses showed that efficacy trended consistently in favor of patients who received Adcetris + AVD compared with patients who received ABVD for most subgroups, as summarized in Figure 11. (See Figure 11.)
Click on icon to see table/diagram/image
Approximately one-third fewer patients treated with Adcetris + AVD received subsequent salvage chemotherapy (n=66) and high-dose chemotherapy and transplant (n=36) compared with those treated with ABVD (n=99 and n=54, respectively).
The European Organization for Research and Treatment of Cancer Quality of Life 30-Item Questionnaire (EORTC-QLQ-C30) showed no clinically meaningful difference between the two arms.
Post-hoc Subgroup Analyses: Subgroups including patients with Stage IV disease and extranodal sites ≥ 1 experienced a greater clinical benefit based on mPFS compared with the overall ITT population. The results from post-hoc analyses in patients with Stage IV disease and extranodal involvement are shown in Table 11. (See Table 11.)
Click on icon to see table/diagram/image
Study SGN35-014: The efficacy of ADCETRIS in combination with chemotherapy for the treatment of adult patients with previously untreated, CD30-expressing PTCL was evaluated in a multicenter, randomized, double-blind, double-dummy, actively controlled trial. For enrollment, the trial required CD30 expression ≥10% per immunohistochemistry. The trial excluded patients with primary cutaneous CD30-positive T-cell lymphoproliferative disorders and lymphomas. The trial required hepatic transaminases ≤3 times ULN, total bilirubin ≤1.5 times ULN, and serum creatinine ≤2 times ULN.
Of the 452 total patients, 226 patients were randomized to the ADCETRIS + CHP arm and 226 patients were randomized to the CHOP arm. Patients in both treatment arms were treated intravenously on Day 1 of each 21-day cycle for 6 to 8 cycles; prednisone was administered orally on Days 1-5. Dosing in each treatment arm was administered according to the following: ADCETRIS + CHP arm: ADCETRIS 1.8 mg/kg over 30 minutes, cyclophosphamide 750 mg/m
2, doxorubicin 50 mg/m
2, and prednisone 100 mg orally; CHOP arm: cyclophosphamide 750 mg/m
2, doxorubicin 50 mg/m
2, vincristine 1.4 mg/m
2, and prednisone 100 mg orally.
The median age was 58 years (range: 18 to 85), 63% were male, 62% were White, 22% were Asian, and 78% had an ECOG performance status of 0-1. Of the 452 patients enrolled, the disease subtypes included patients with systemic ALCL [70%; 48% anaplastic lymphoma kinase (ALK) negative and 22% ALK positive], PTCL not otherwise specified (16%), angioimmunoblastic T-cell lymphoma (12%), adult T-cell leukemia/lymphoma (2%), and enteropathy-associated T-cell lymphoma (<1%).
Most patients had Stage III or IV disease (81%) and a baseline international prognostic index of 2 or 3 (63%).
During randomized treatment, on the ADCETRIS + CHP arm, 70% of patients received 6 cycles and 18% of patients received 8 cycles. On the CHOP arm, 62% of patients received 6 cycles and 19% received 8 cycles.
Efficacy was based on IRF-assessed PFS, which was defined as time from randomization to progression, death due to any cause, or receipt of subsequent anticancer chemotherapy to treat residual or progressive disease. Other efficacy endpoints included PFS in patients with systemic ALCL, overall survival, complete response rate, and overall response rate. Efficacy results are summarized in Table 12: Kaplan-Meier curves for PFS and overall survival are presented in Figure 12 and Figure 13, respectively. (See Table 12, Figures 12 and 13.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Median overall survival was not reached in either treatment arm.
Pharmacokinetics: General Introduction: The pharmacokinetics of Adcetris were evaluated in phase 1 studies and in a population pharmacokinetic analysis of data from 314 patients.
Absorption and Bioavailability: Monotherapy: The serum pharmacokinetics of ADC following an intravenous dose of Adcetris were similar to other antibody products. Maximum concentrations were typically observed at the end of infusion or the sampling time point closest to the end of infusion. A multiexponential decline in ADC serum concentrations was observed with a terminal half-life of approximately 4 to 6 days. Exposures were approximately dose proportional. After multiple-dose administration of Adcetris, ADC steady-state was achieved by 21 days, consistent with the terminal half-life estimate. Minimal to no accumulation of ADC was observed with multiple doses at the every 3-week schedule.
The elimination of MMAE was limited by its rate of release from ADC. The time to maximum concentration ranged from approximately 1 to 3 days after each infusion. MMAE exposures decreased after multiple doses of Adcetris with approximately 50% to 80% of the exposure of the first dose being observed at subsequent doses.
Combination Therapy: The pharmacokinetics of Adcetris in combination with doxorubicin, vinblastine, and dacarbazine (AVD) were evaluated in a single phase 3 study in 661 patients (C25003). Population pharmacokinetic analysis indicated that the pharmacokinetics of Adcetris in combination with AVD were consistent with that in monotherapy.
After multiple-dose, IV infusion of 1.2 mg/kg Adcetris every two weeks, maximal serum concentrations of ADC were observed near the end of the infusion and elimination exhibited a multiexponential decline with a t
½z of approximately 4 to 5 days. Maximal plasma concentrations of MMAE were observed approximately 2 days after the end of infusion, and exhibited a mono-exponential decline with a t
½z of approximately 3 to 4 days.
After multiple-dose, IV infusion of 1.2 mg/kg Adcetris every two weeks, steady-state trough concentrations of ADC and MMAE were achieved by Cycle 3. Once steady-state was achieved, the pharmacokinetics (PK) of ADC did not appear to change with time. ADC accumulation (as assessed by AUC
14D between Cycle 1 and Cycle 3) was 1.27-fold. The exposure of MMAE (as assessed by AUC
14D between Cycle 1 and Cycle 3) appeared to decrease with time by approximately 50%.
The pharmacokinetics of Adcetris in combination with CHP were evaluated in a single phase 3 study in 223 patients (SGN35-014). After multiple-dose IV infusion of 1.8 mg/kg Adcetris every 3 weeks, the pharmacokinetics of ADC and MMAE were similar to those of monotherapy.
Distribution: In vitro, the binding of MMAE to human serum plasma proteins ranged from 68-82%. MMAE is not likely to displace or to be displaced by highly protein-bound drugs. In vitro, MMAE was a substrate of P-gp and was not a potent inhibitor of P-gp.
In humans, the mean steady state volume of distribution was approximately 6-10 L for ADC.
Metabolism: In vivo data in animals and humans suggests that only a small fraction of MMAE released from Adcetris is metabolized. In vitro data indicate that the MMAE metabolism that occurs is primarily via oxidation by CYP3A4/5. In vitro studies using human liver microsomes indicate that MMAE inhibits CYP3A4/5 but not other isoforms. MMAE did not induce any major CYP450 enzymes in primary cultures of human hepatocytes.
Elimination: An excretion study was undertaken in patients who received a dose of 1.8 mg/kg of Adcetris (brentuximab vedotin). Approximately 24% of the total MMAE administered as part of the ADC during an Adcetris infusion was recovered in both urine and feces over a 1-week period. Of the recovered MMAE, approximately 72% was recovered in the feces and the majority of the excreted MMAE was unchanged. A lesser amount of MMAE (28%) was excreted in the urine and the majority was excreted unchanged.
Special Populations: Pediatric: Clinical studies of Adcetris did not include sufficient numbers of subjects below 18 years of age to determine whether they respond differently from older subjects. Safety and efficacy have not been established.
Geriatric: The population pharmacokinetics of brentuximab vedotin as monotherapy were examined from several monotherapy studies, including data from 380 patients up to 87 years old (34 patients ≥65-<75 and 17 patients ≥75 years of age). Additionally, the population pharmacokinetics of brentuximab vedotin in combination with AVD were examined, including data from 661 patients up to 82 years old (42 patients ≥65-<75 and 17 patients ≥75 years of age). The influence of age on pharmacokinetics was investigated in each analysis and it was not a significant covariate.
Renal impairment: A study evaluated the pharmacokinetics of Adcetris and MMAE after the administration of 1.2 mg/kg of Adcetris to patients with mild (n=4), moderate (n=3) and severe (n=3) renal impairment. Compared to patients with normal renal function, MMAE exposure increased approximately 1.9-fold in patients with severe renal impairment.
Hepatic impairment: A study evaluated the pharmacokinetics of Adcetris and MMAE after the administration of 1.2 mg/kg of Adcetris to patients with mild (Child-Pugh A; n=1), moderate (Child-Pugh B; n=5) and severe (Child-Pugh C; n=1) hepatic impairment. Compared to patients with normal hepatic function, MMAE exposure increased approximately 2.3-fold in patients with hepatic impairment.
Toxicology: NONCLINICAL TOXICOLOGY: Carcinogenesis, Mutagenesis, Impairment of Fertility: Carcinogenicity: Carcinogenicity studies with Adcetris (brentuximab vedotin) or MMAE have not been conducted.
Mutagenicity: MMAE was negative for mutagenicity in the bacterial reverse mutation assay (Ames test) and the mouse lymphoma forward mutation assay. The in vivo rat bone marrow micronucleus study revealed aneugenic rather than clastogenic micronuclear formation. These results were consistent with the pharmacological effect of MMAE on the mitotic apparatus (disruption of the microtubule network) in cells.
Impairment of Fertility: The effects of Adcetris on human male and female fertility have not been studied. However, results of repeat-dose toxicity studies in rats indicate the potential for Adcetris to impair male reproductive function and fertility. Testicular atrophy and degeneration were observed in a 4-week rat study when Adcetris was given weekly at intravenous doses of 5 or 10 mg/kg.
These changes were partially reversible following a 16-week treatment-free period.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image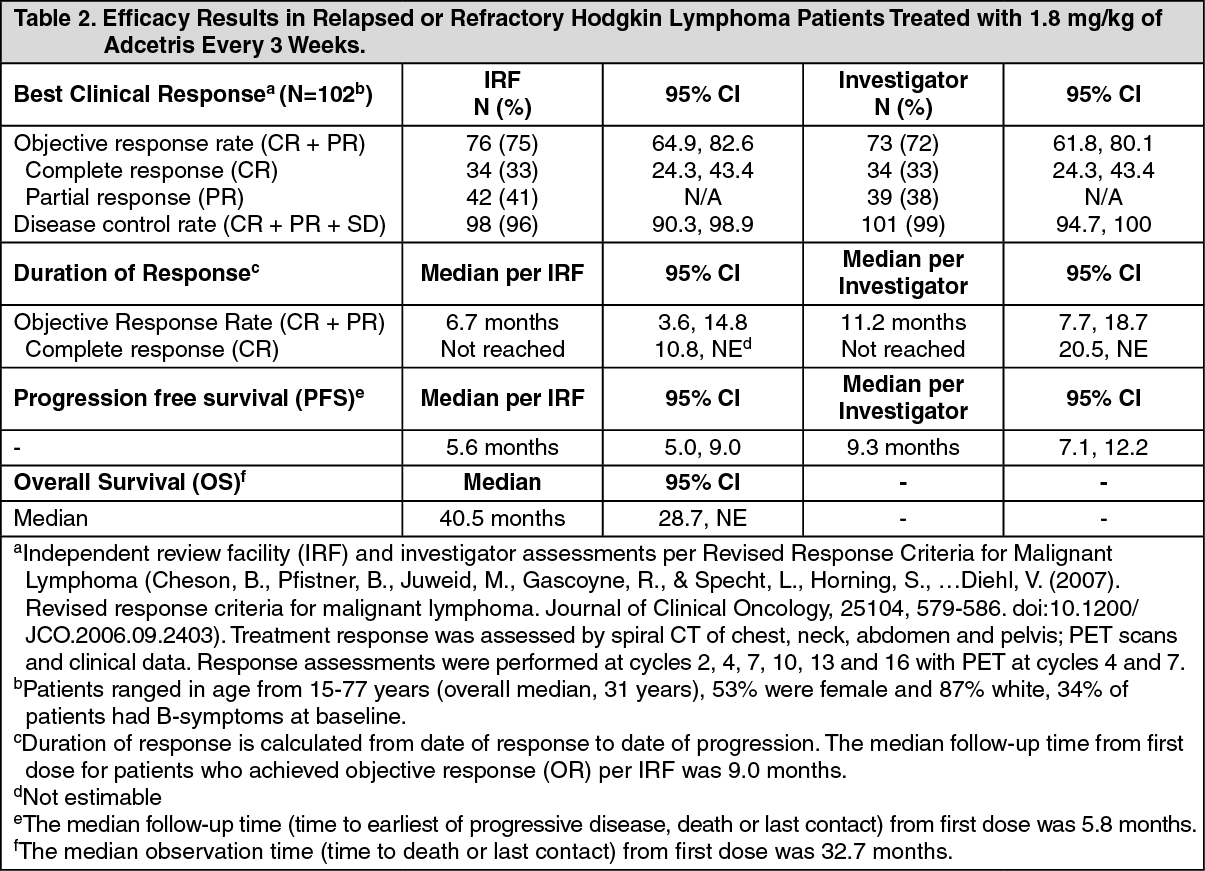 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image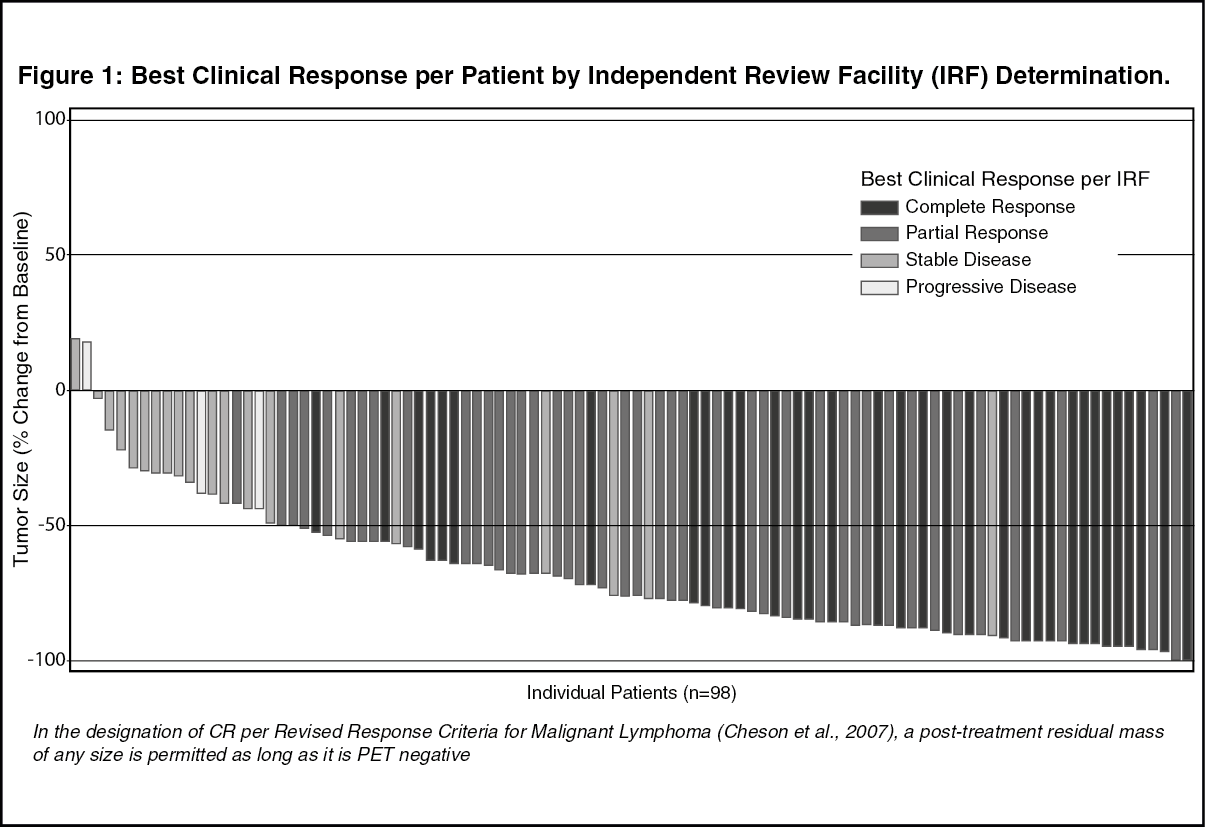 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image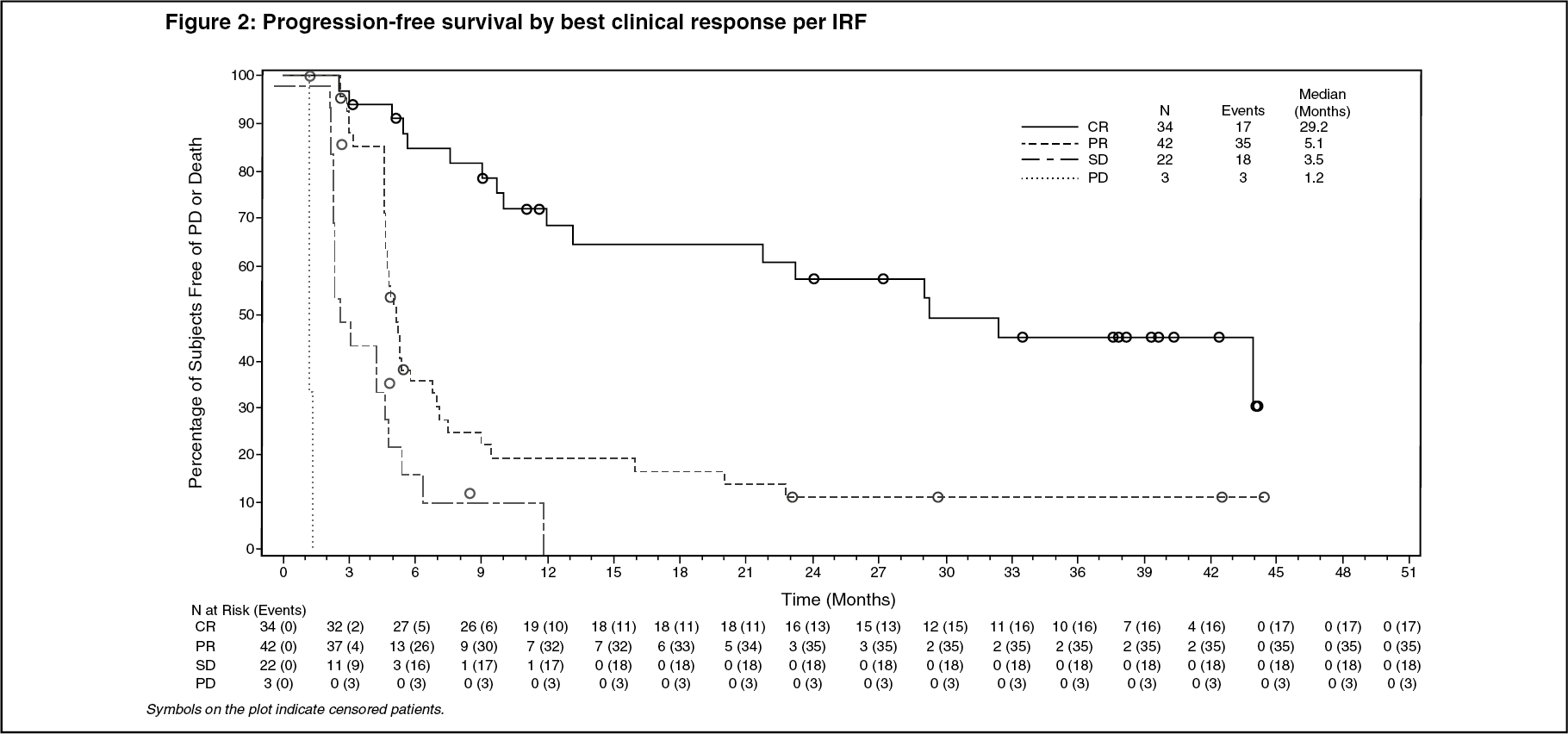 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image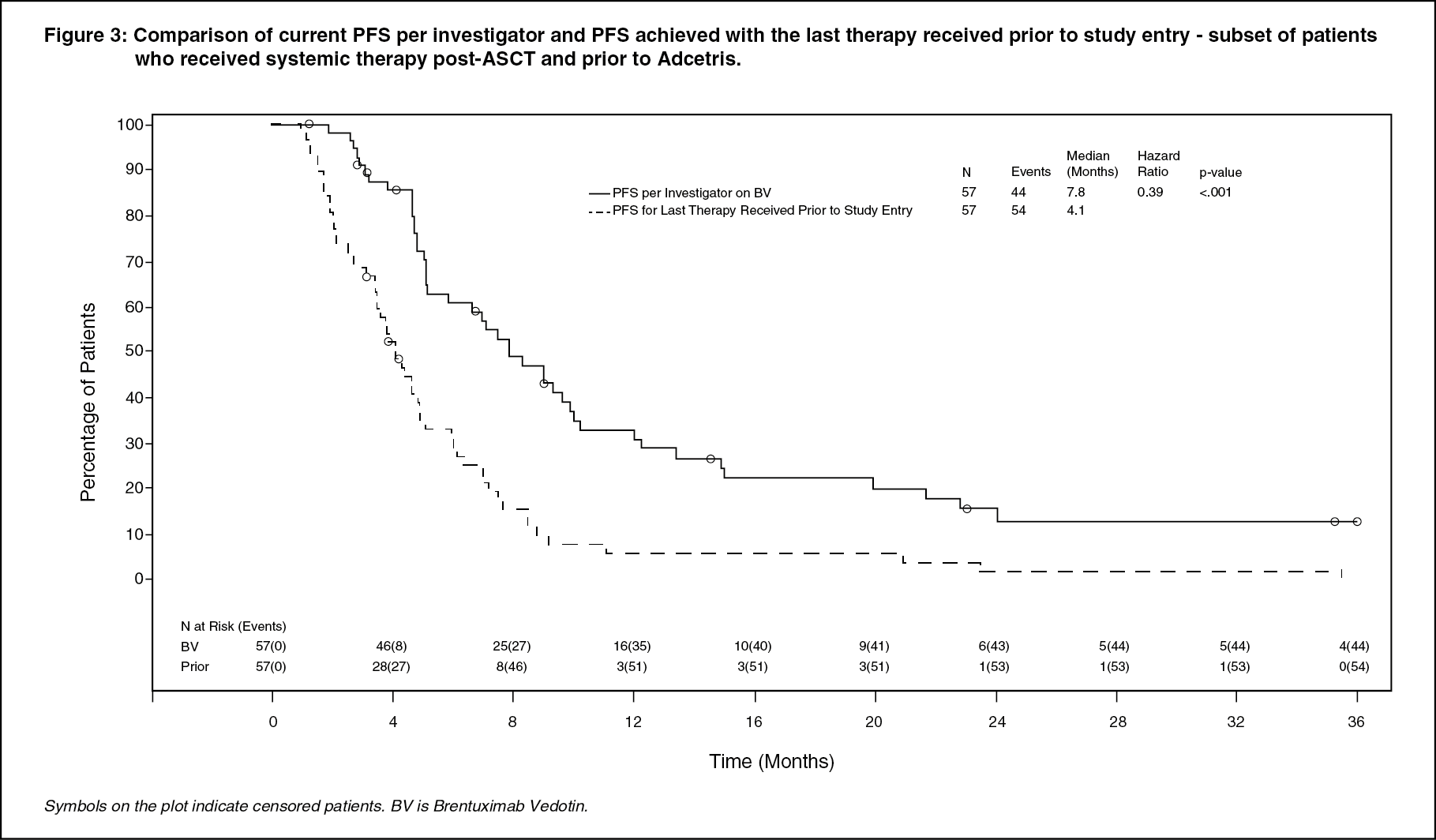 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image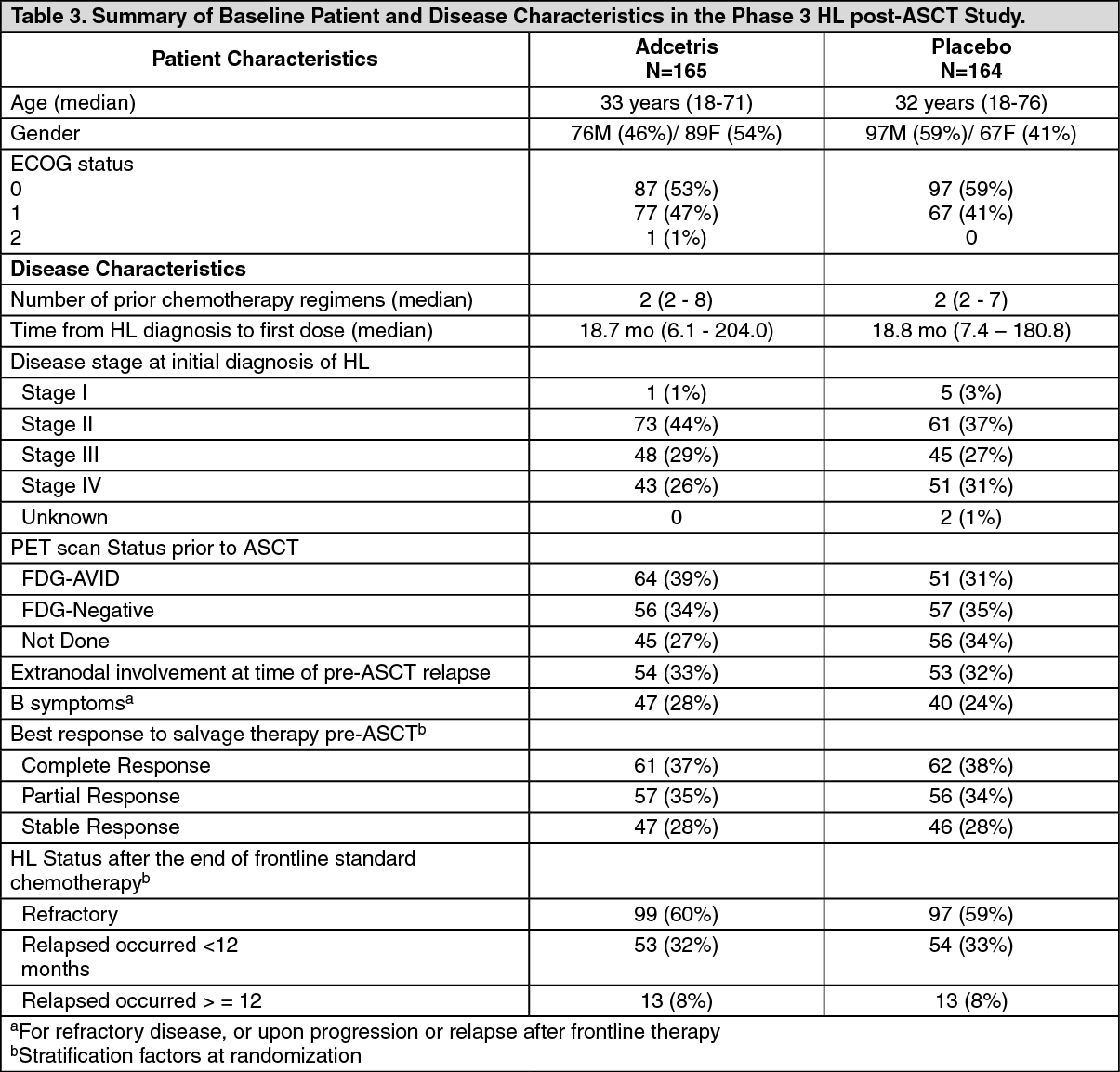 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image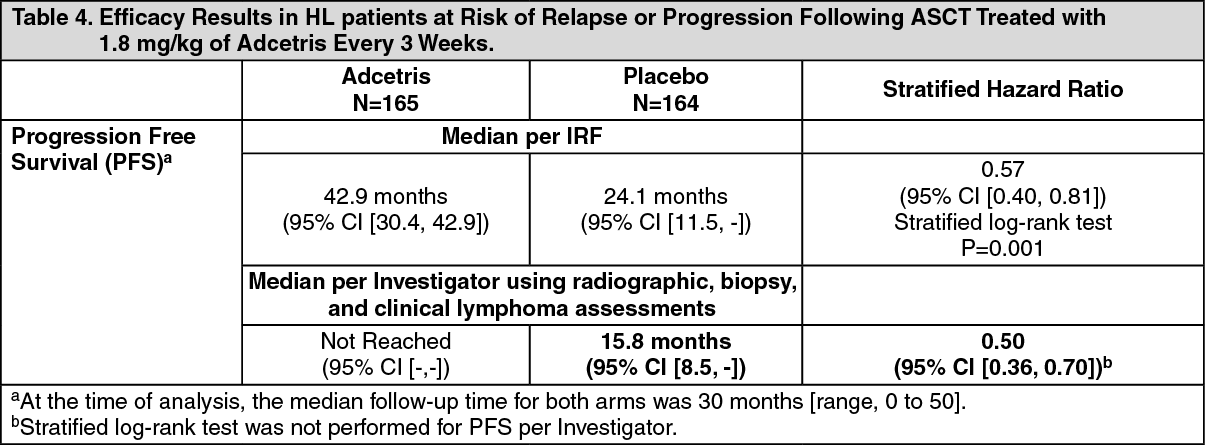 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image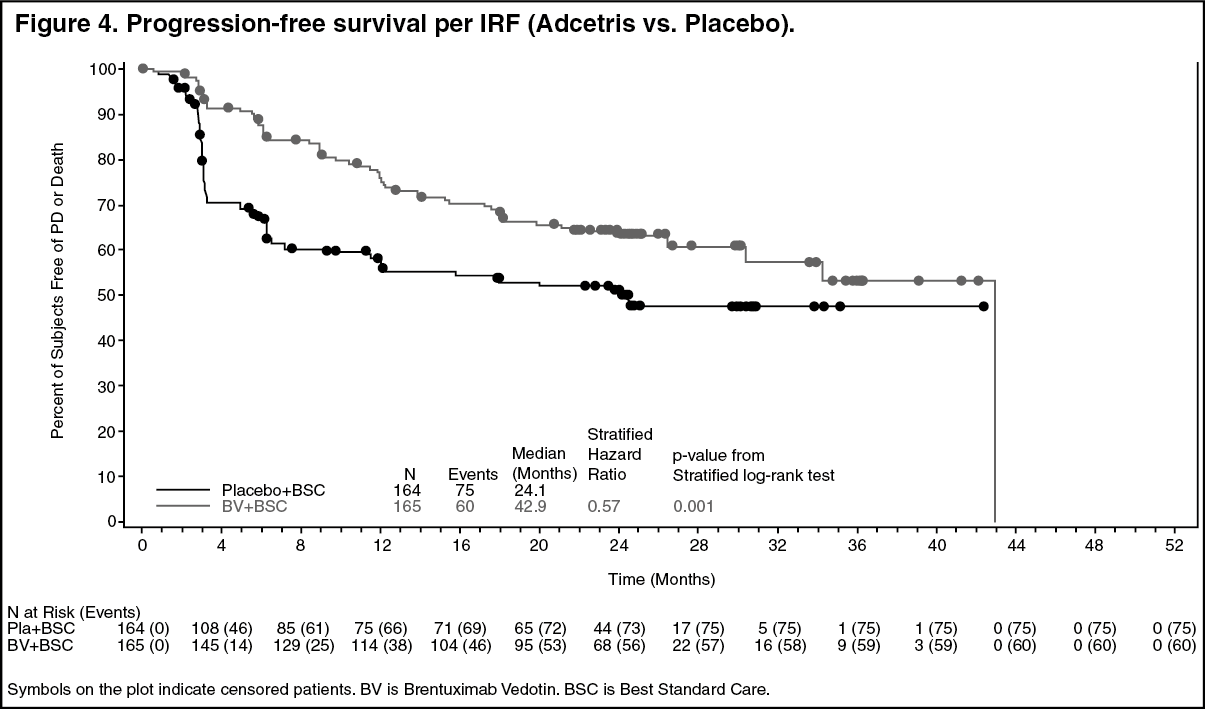 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image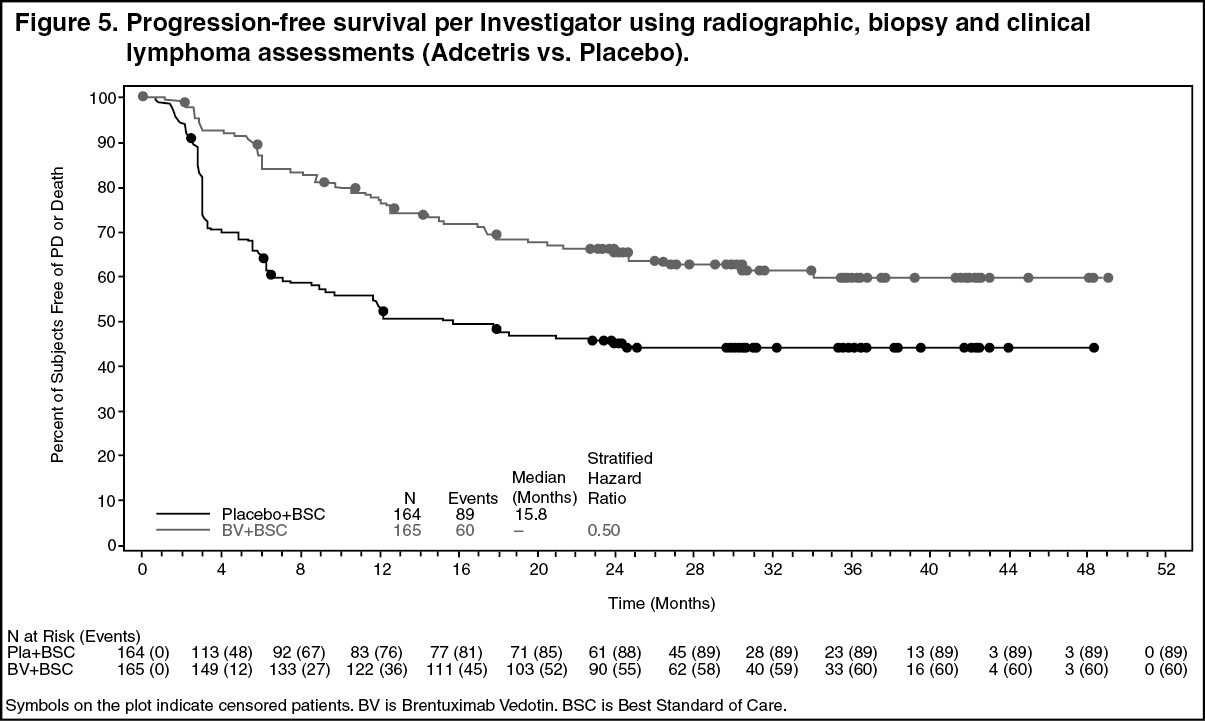 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image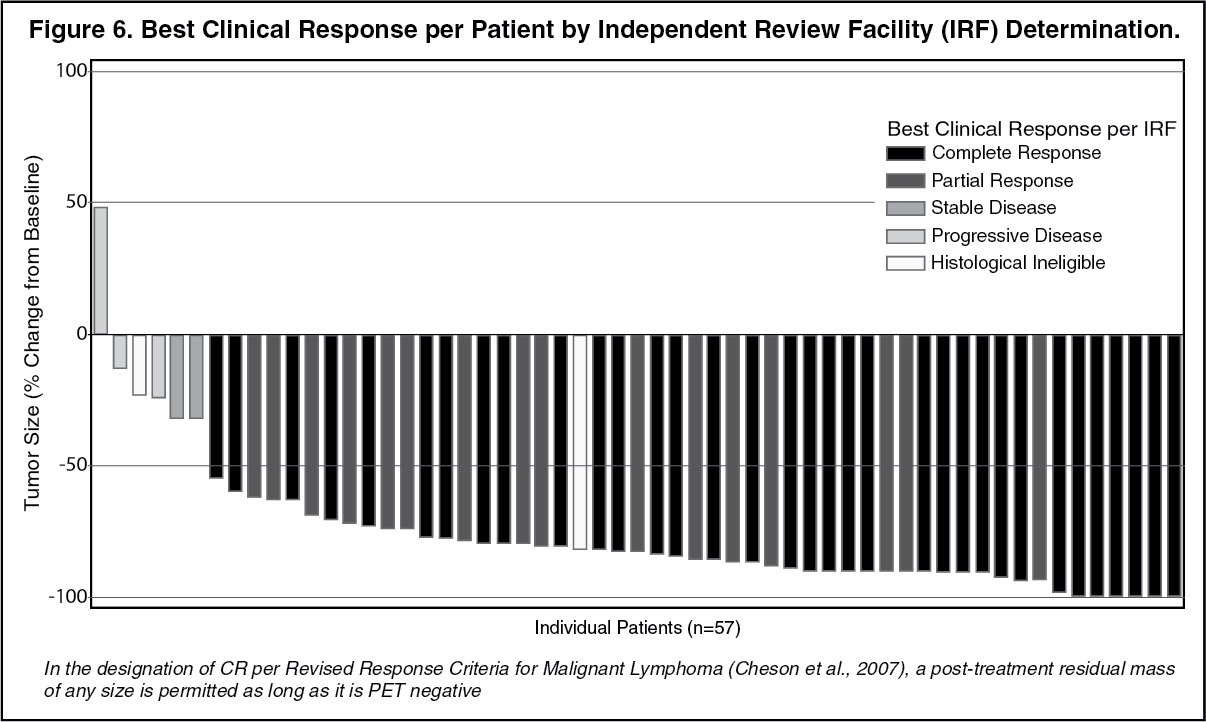 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image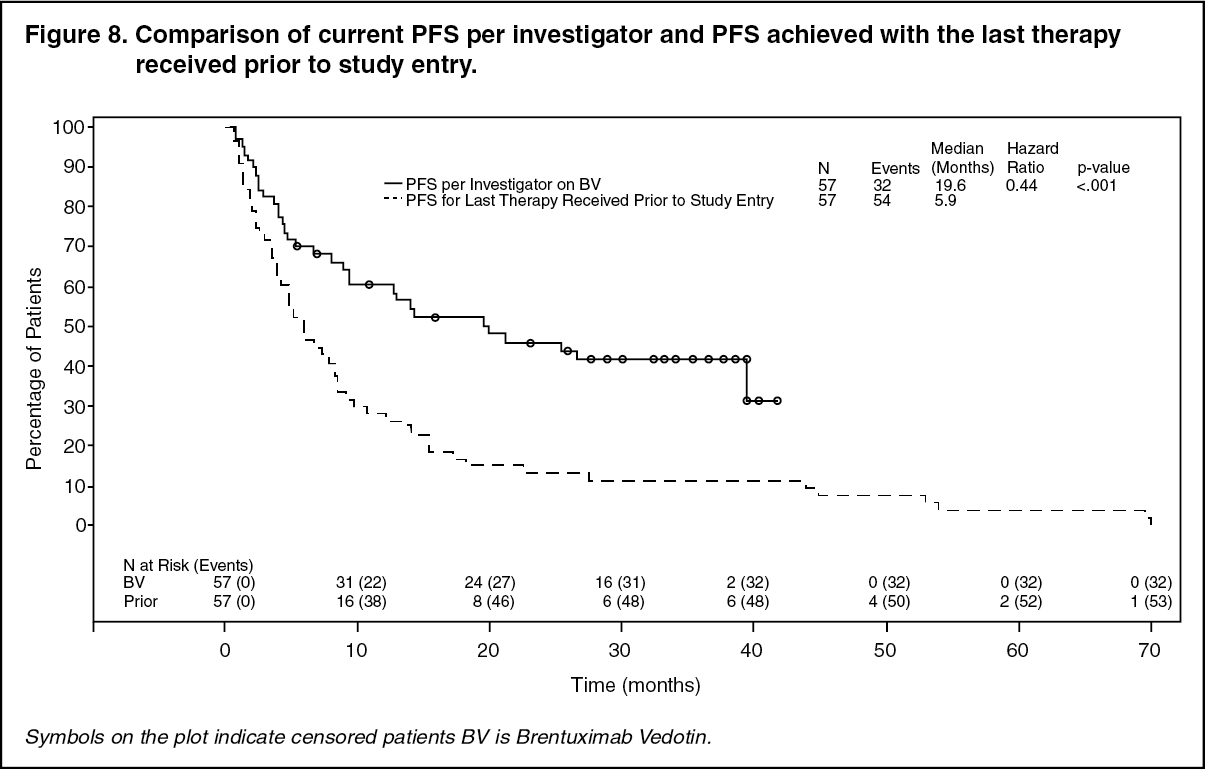 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image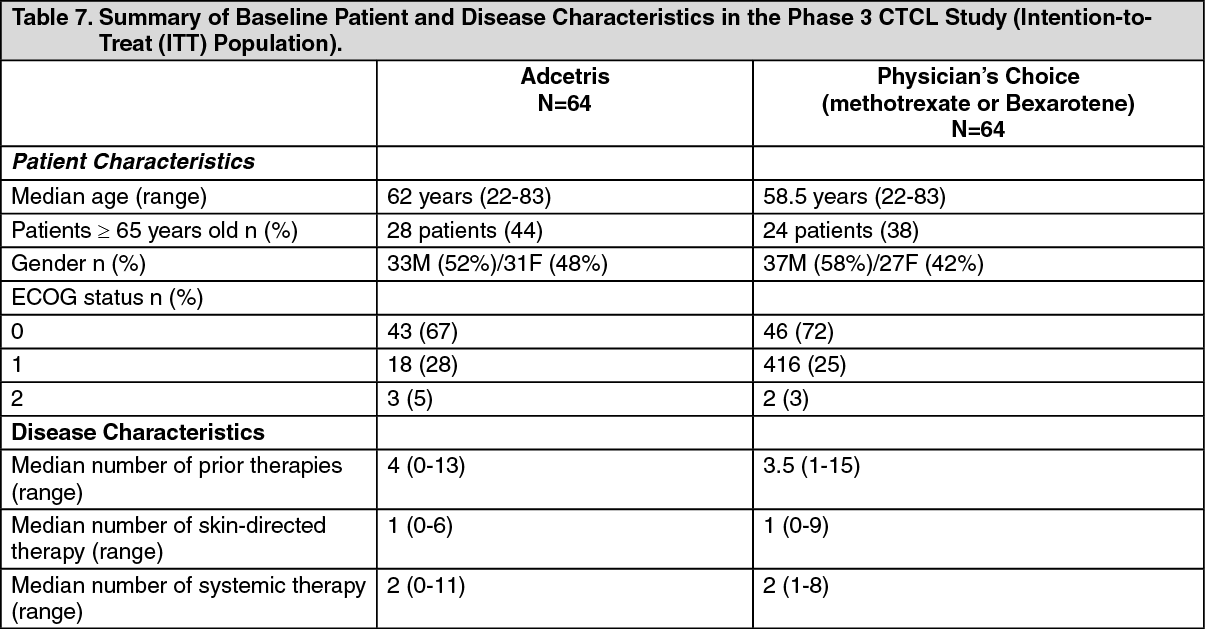 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image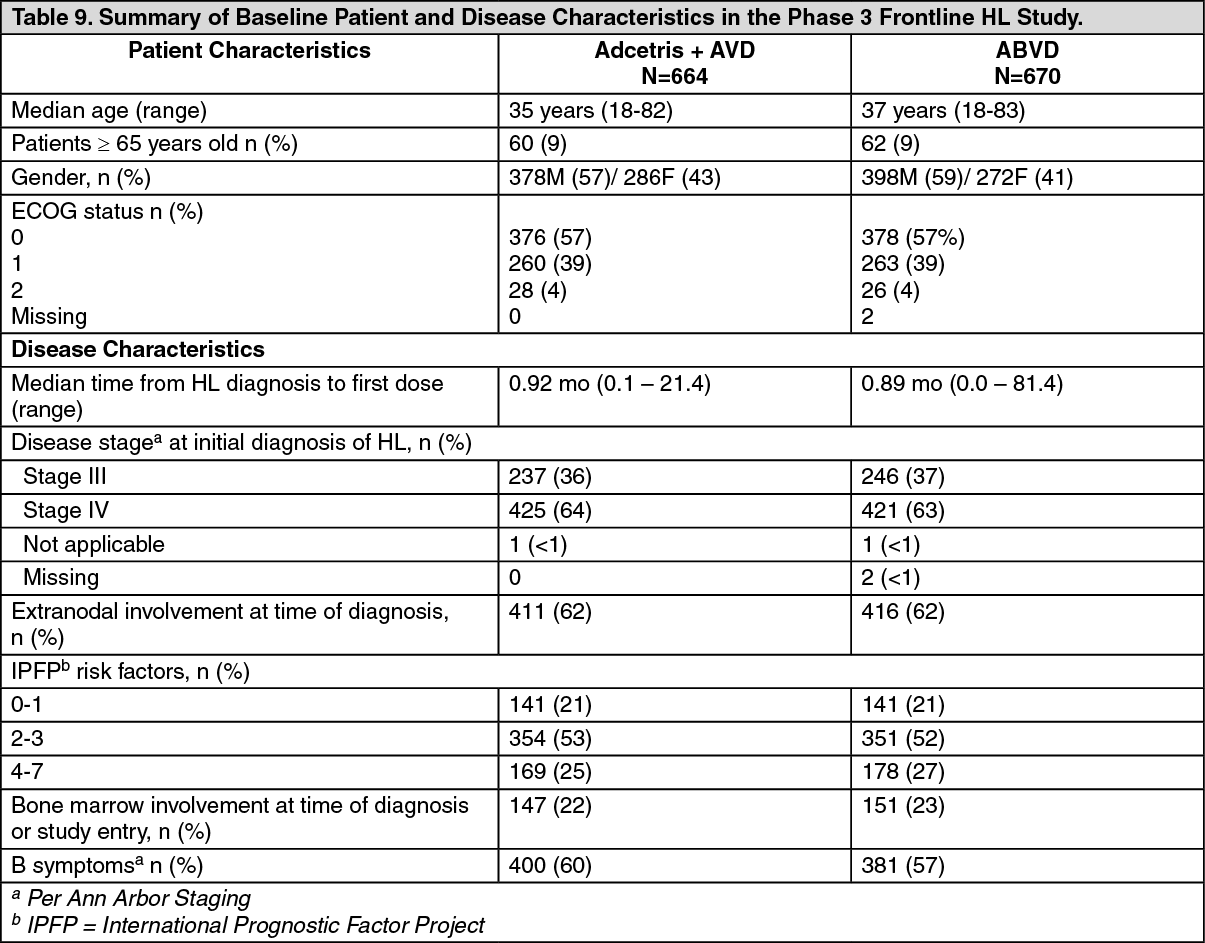 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image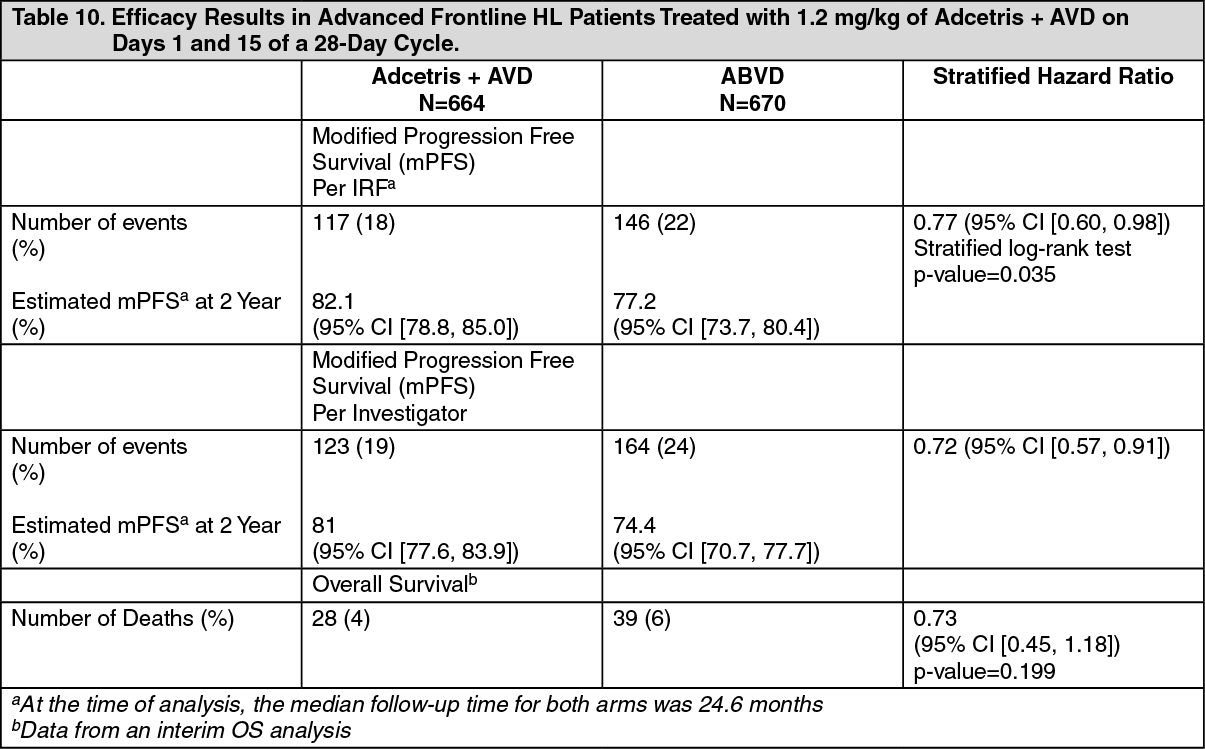 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image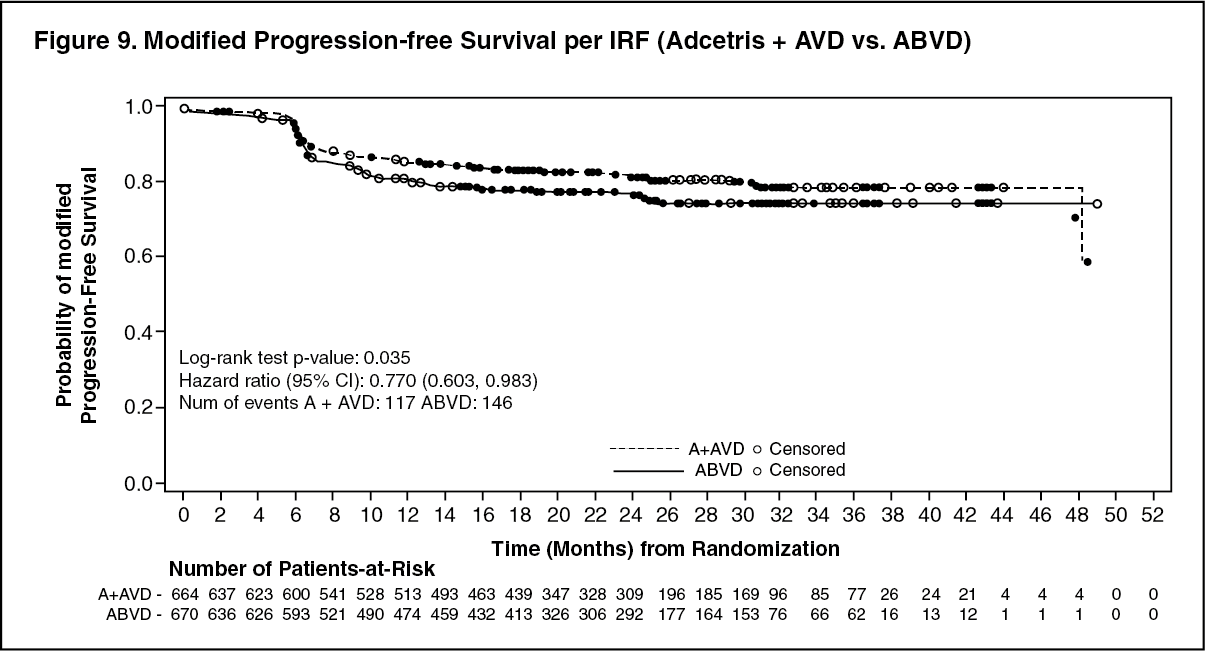 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image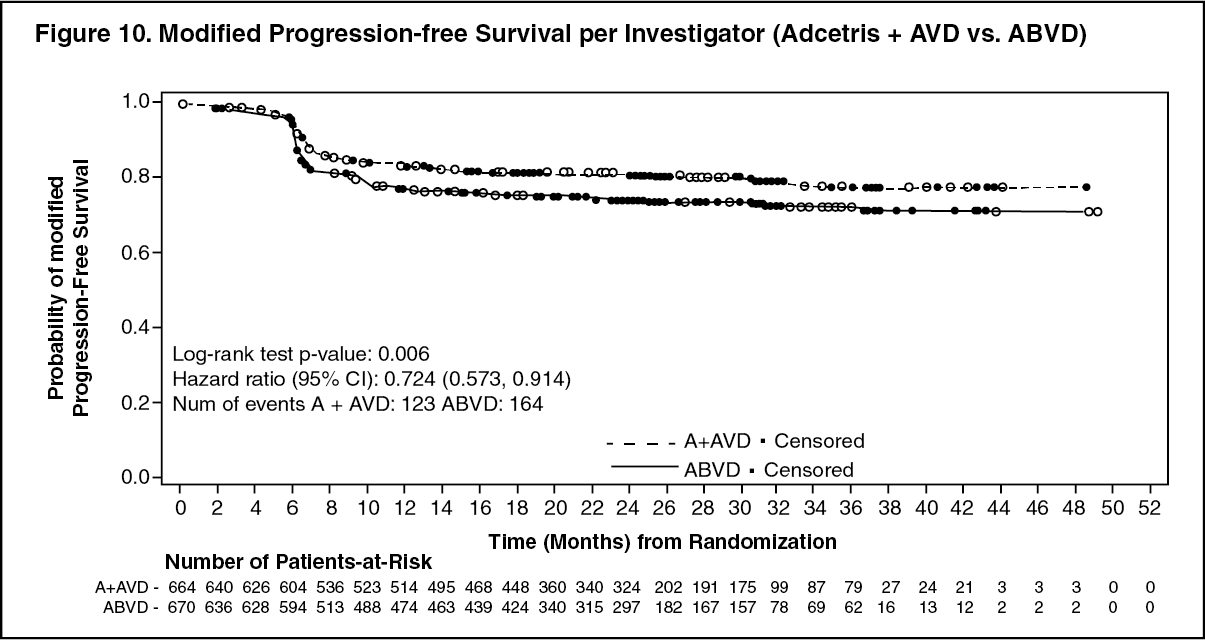 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image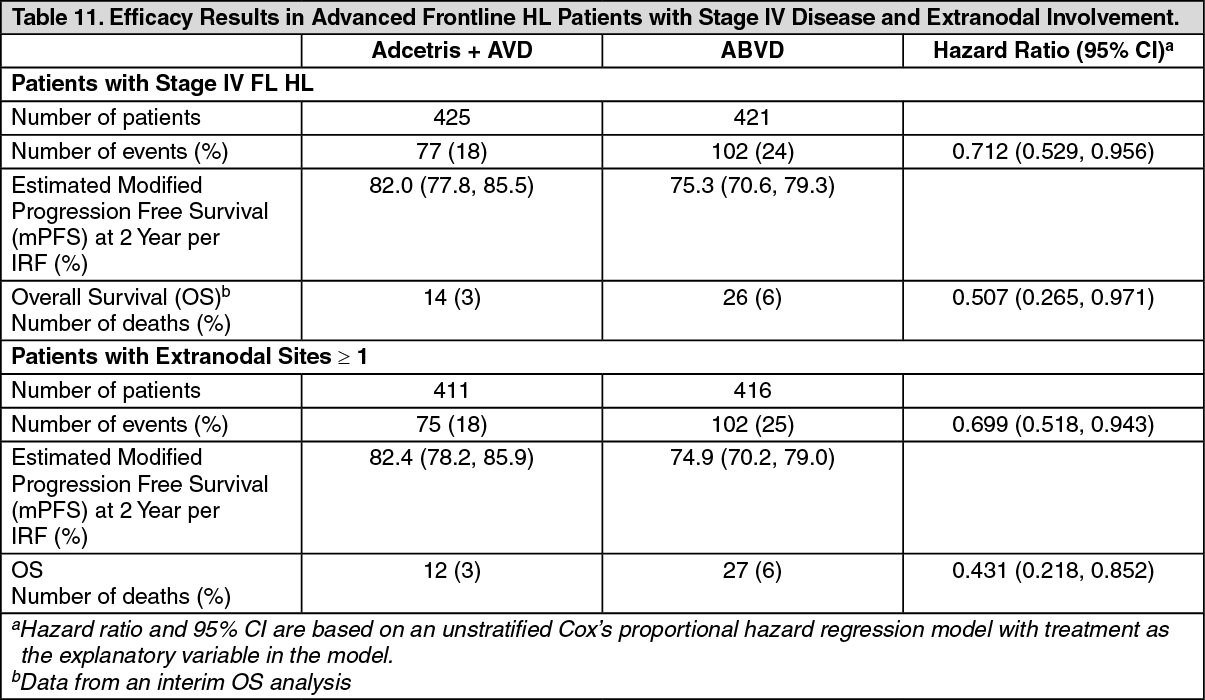 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image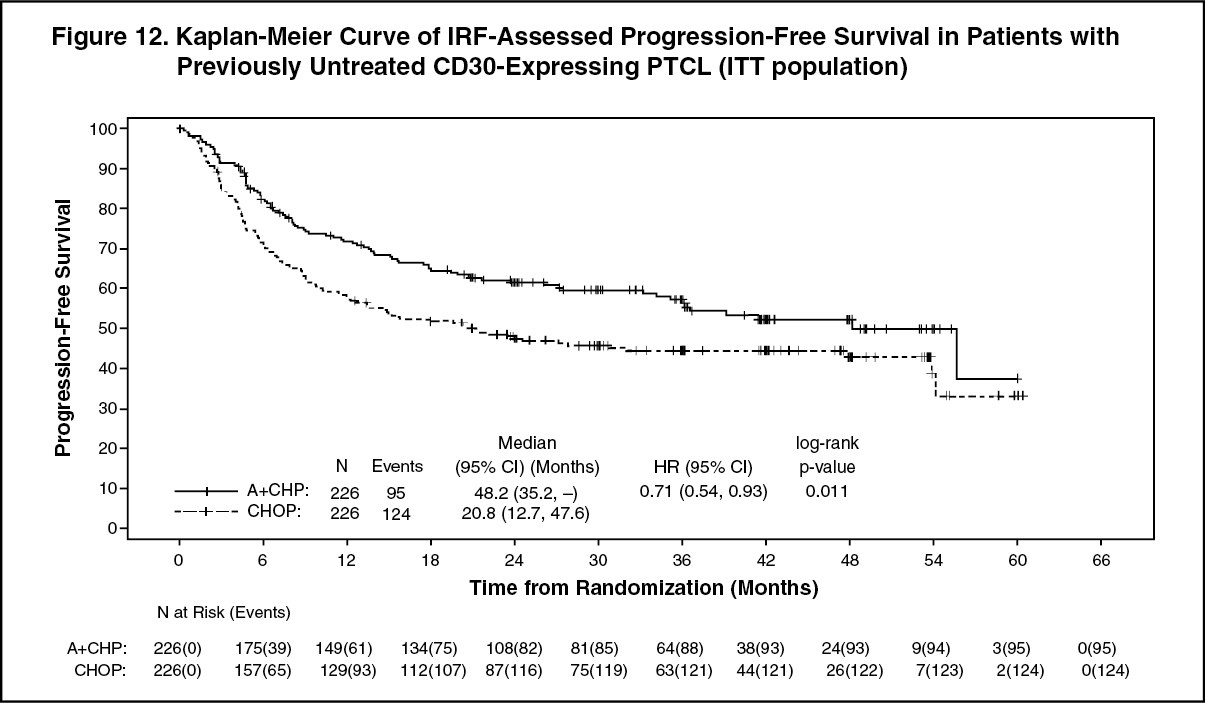 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image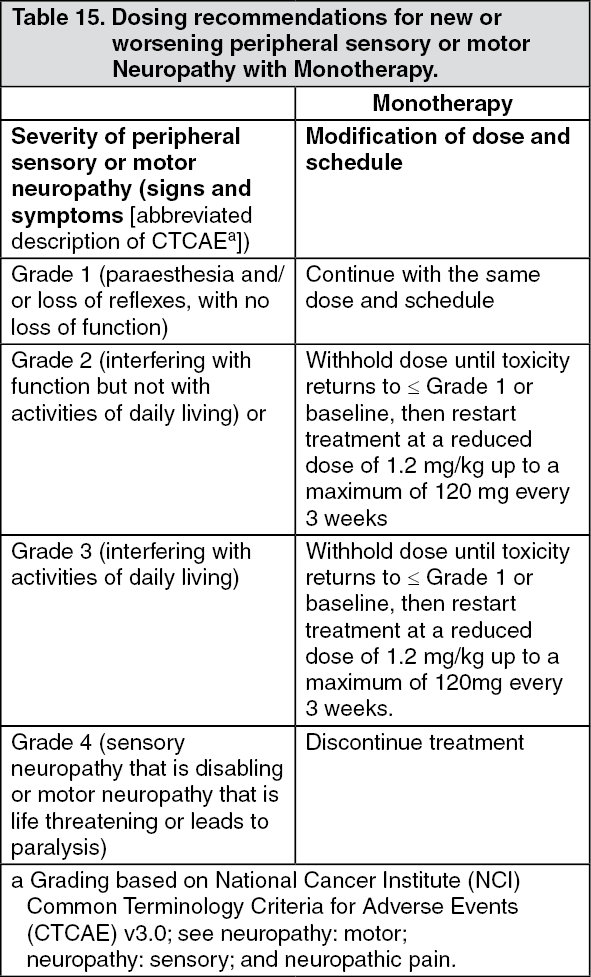 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image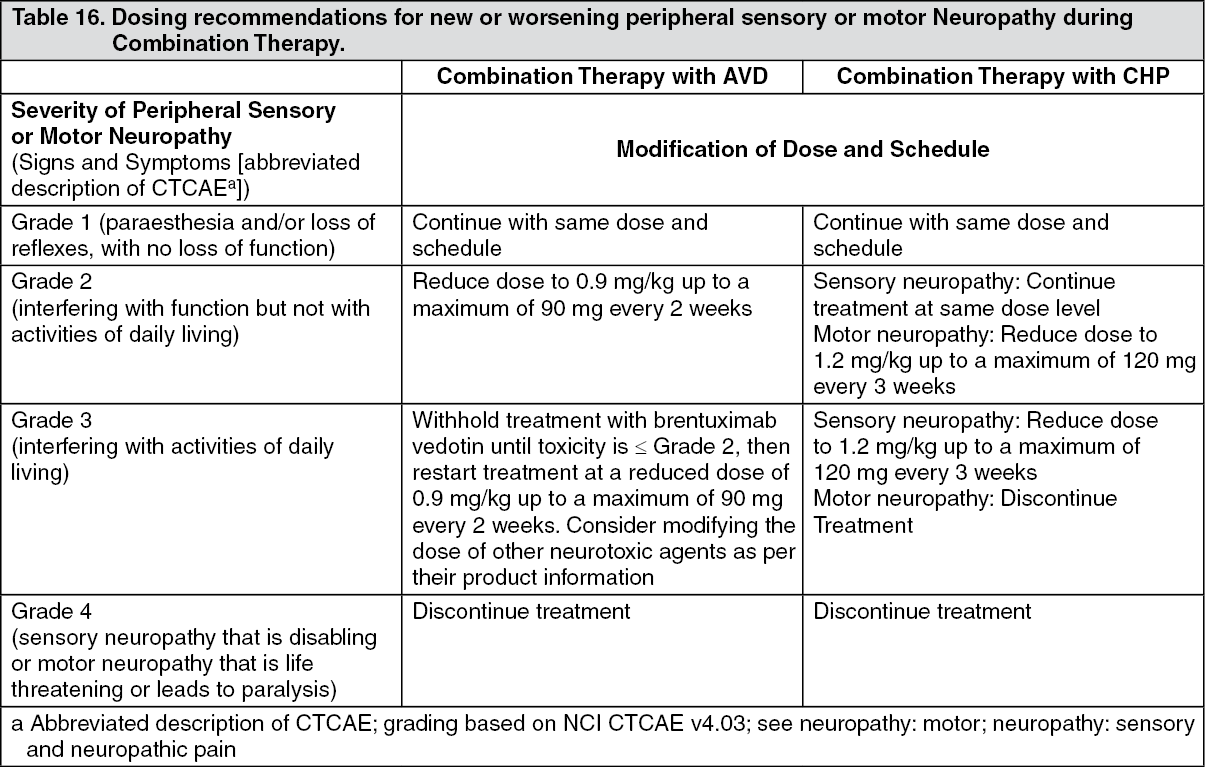 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image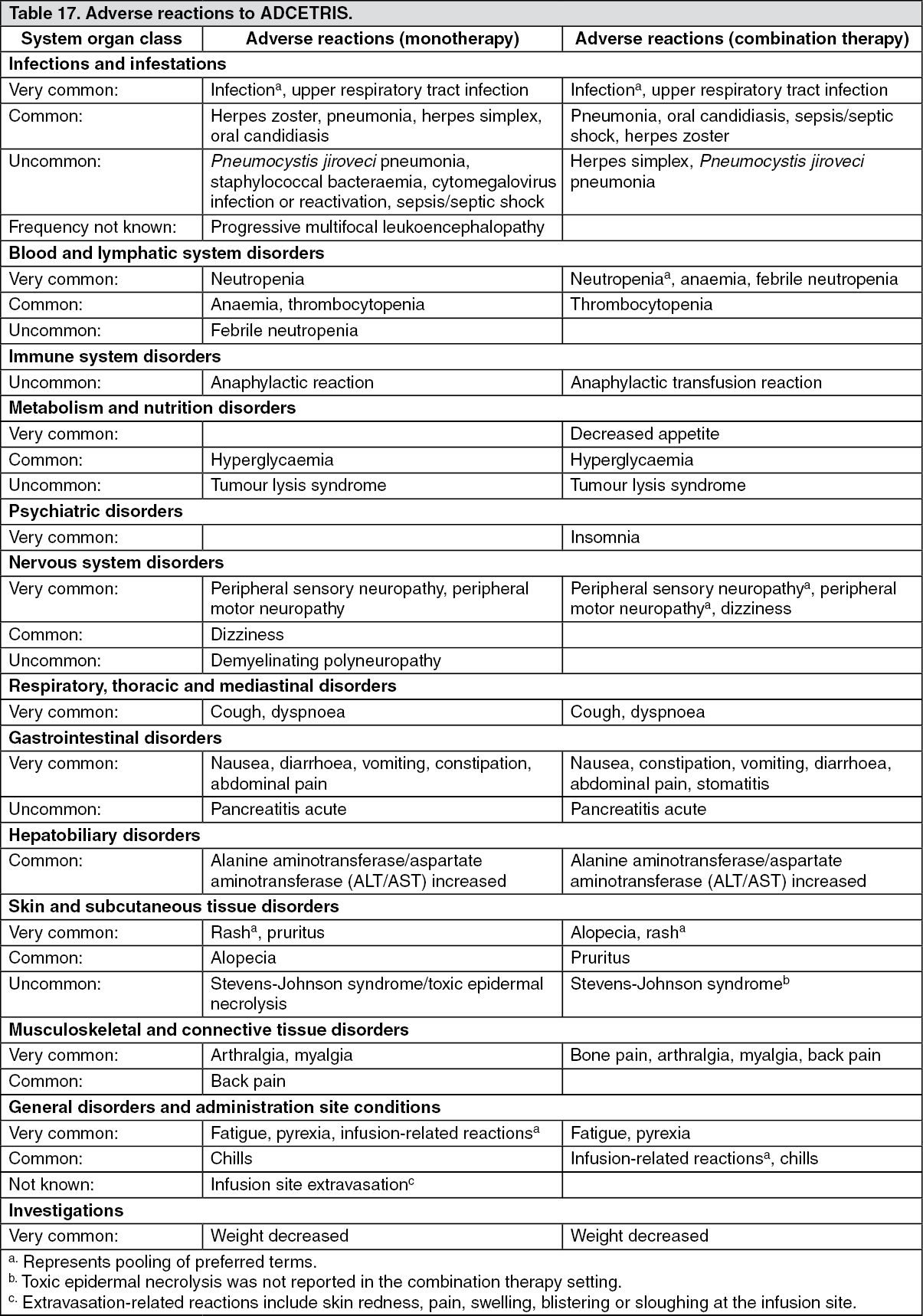 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
