


 Sign Out
Sign Out

Monotherapy: In the pooled dataset of Adcetris as monotherapy across HL, sALCL and CTCL studies (SG035-0003, SG035-0004, SGN35-005, SGN35-006, C25001 and C25007, see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions), the most frequent adverse reactions (≥10%) were infections, peripheral sensory neuropathy, fatigue, nausea, diarrhea, pyrexia, upper respiratory tract infection, neutropenia, rash, cough, vomiting, arthralgia, peripheral motor neuropathy, infusion-related reactions, pruritus, constipation, dyspnea, weight decreased, myalgia and abdominal pain.
Serious adverse drug reactions occurred in 12% of patients. The frequency of unique serious adverse drug reactions was ≤ 1%.
Adverse events led to treatment discontinuation in 24% of patients receiving ADCETRIS.
The safety data in patients retreated with ADCETRIS (SGN35-006, see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions) were consistent with those observed in the combined pivotal phase 2 studies, with the exception of peripheral motor neuropathy, which had a higher incidence (28% vs. 9% in the pivotal phase 2 studies) and was primarily Grade 2. Patients also had a higher incidence of arthralgia, Grade 3 anaemia, and back pain compared to patients observed in the combined pivotal phase 2 studies.
The safety data in patients with relapsed or refractory HL who had not received an autologous stem cell transplant and were treated with the recommended dose of 1.8 mg/kg every three weeks in a single-arm phase 4 study (n = 60), the phase 1 dose escalation and clinical pharmacology studies (n = 15 patients) and in the NPP (n = 26 patients) (see Pharmacology: Pharmacodynamics: CLINICAL STUDIES under Actions) were consistent with the safety profile of the pivotal clinical studies.
Combination therapy: For safety information of chemotherapy agents given in combination with ADCETRIS (doxorubicin, vinblastine and dacarbazine (AVD) or cyclophosphamide, doxorubicin and prednisone (CHP)), refer to their summary of product characteristics.
In the studies of ADCETRIS as combination therapy in 662 patients with previously untreated advanced HL (C25003) and 223 patients with previously untreated CD30+ peripheral T-cell lymphoma (PTCL) (SGN35-014), the most common adverse reactions (≥ 10%) were: infections, neutropenia, peripheral sensory neuropathy, nausea, constipation, vomiting, diarrhoea, fatigue, pyrexia, alopecia, anaemia, weight decreased, stomatitis, febrile neutropenia, abdominal pain, decreased appetite, insomnia, bone pain, rash, cough, dyspnoea, arthralgia, myalgia, back pain, peripheral motor neuropathy, upper respiratory tract infection, and dizziness.
In patients receiving ADCETRIS combination therapy, serious adverse reactions occurred in 34% of patients. Serious adverse reactions occurring in ≥ 3% of patients included febrile neutropenia (15%), pyrexia (5%), and neutropenia (3%).
Adverse events led to treatment discontinuation in 10% of patients. Adverse events that led to treatment discontinuation in ≥ 2% of patients included peripheral sensory neuropathy, and peripheral neuropathy.
Tabulated list of adverse reactions: Adverse reactions for ADCETRIS are listed by MedDRA System Organ Class and Preferred Term (see Table 17). Within each System Organ Class, adverse reactions are listed under frequency categories of: Very common (≥ 1/10); Common (≥ 1/100 to < 1/10); Uncommon (≥ 1/1,000 to < 1/100); Rare (≥ 1/10,000 to < 1/1,000); Very rare (< 1/10,000); not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in the order of decreasing seriousness. (See Table 17.)
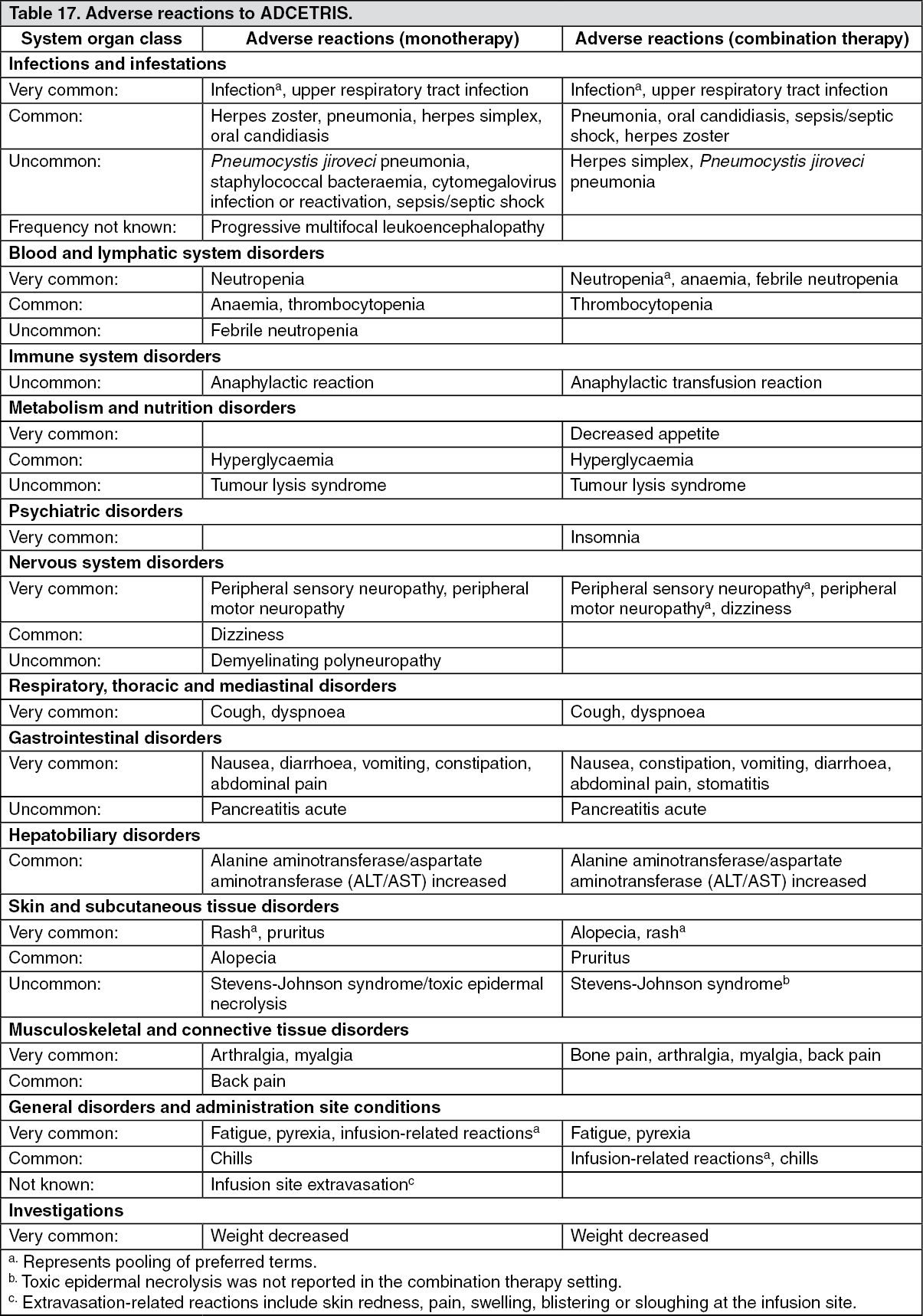 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Neutropenia and febrile neutropenia: Monotherapy: In clinical trials, neutropenia led to dose delays in 14% of patients. Grade 3 neutropenia was reported in 13% and Grade 4 neutropenia was reported in 5% of patients. No patients required dose reduction or discontinued treatment for neutropenia.
Severe and prolonged (≥ 1 week) neutropenia can occur with this treatment which may increase the risk of patients developing serious infections. Febrile neutropenia reported in < 1% of the patients (see DOSAGE & ADMINISTRATION).
In the pivotal phase 2 population (SG035-0003 and SG035-0004), the median duration of Grade 3 or Grade 4 neutropenia was limited (1 week); 2% of patients had Grade 4 neutropenia that lasted ≥ 7 days. Less than half of the patients in the pivotal phase 2 population with Grade 3 or Grade 4 neutropenia had temporally associated infections, and the majority of temporally associated infections were Grade 1 or Grade 2.
Combination therapy: In the clinical trials of ADCETRIS as combination therapy, neutropenia led to dose delays in 19% of patients. Grade 3 neutropenia was reported in 17% and Grade 4 neutropenia was reported in 41% of patients. Two percent of patients required dose reduction and < 1% discontinued one of more of the study drugs due to neutropenia.
Febrile neutropenia was reported in 20% of the patients who did not receive primary prophylaxis with G-CSF (see DOSAGE & ADMINISTRATION). The frequency of febrile neutropenia was 13% in patients who received primary prophylaxis with G-CSF.
Serious infections and opportunistic infections: Monotherapy: In clinical trials, serious infections and opportunistic infections occurred in 10% of patients, sepsis or septic shock occurred in < 1% of the patients. The most commonly reported opportunistic infections were herpes zoster and herpes simplex.
Combination therapy: In the clinical trials of ADCETRIS as combination therapy, serious infections including opportunistic infections occurred in 15% of patients; sepsis, neutropenic sepsis, septic shock or bacteraemia occurred in 4% of the patients. The most commonly reported opportunistic infections were herpes viral infections.
Peripheral neuropathy: Monotherapy: In clinical trials treatment emergent neuropathy occurred in 59% of the population, peripheral motor neuropathy occurred in 14% of patients. Peripheral neuropathy led to treatment discontinuation in 15%, dose reductions in 15%, and dose delays in 17% of patients. For patients who experienced peripheral neuropathy the median time of onset of peripheral neuropathy was 12 weeks. The median duration of treatment for patients who discontinued due to peripheral neuropathy was 12 cycles.
Among patients who experienced peripheral neuropathy in the pivotal phase 2 studies (SG035-0003 and SG035-0004) and randomised phase 3 monotherapy studies (SGN35-005 and C25001), the median follow up time from end of treatment until last evaluation ranged from 48.9 to 98 weeks. At the time of last evaluation, most of the patients (82-85%) who experienced peripheral neuropathy had resolution or improvement of their peripheral neuropathy symptoms. The median time from onset to resolution or improvement for all events ranged from 16 to 23.4 weeks.
In patients with relapsed or refractory HL or sALCL who were retreated with ADCETRIS (SGN35-006), the majority of patients (80%) also had improvement or resolution of their peripheral neuropathy symptoms at the time of last evaluation.
Combination therapy: In the clinical trial of ADCETRIS as combination therapy with AVD, treatment emergent neuropathy occurred in 67% of the population; peripheral motor neuropathy occurred in 11% of patients. Peripheral neuropathy led to treatment discontinuation in 7%, dose reductions in 21%, and dose delays in 1% of patients. For patients who experienced peripheral neuropathy the median time of onset of peripheral neuropathy was 8 weeks. Patients who discontinued due to peripheral neuropathy received a median of 8 doses of ADCETRIS+AVD (A+AVD) before discontinuation of one or more agents.
Among patients who experienced peripheral neuropathy, the median follow up time from end of treatment until last evaluation was approximately 91 weeks. At the time of last evaluation, most of the patients (76%) who experienced peripheral neuropathy had resolution or improvement of their peripheral neuropathy symptoms. The median time from onset to resolution or improvement of peripheral neuropathy events was 10 weeks (ranged from 0 weeks to 139 weeks).
In the clinical trial of ADCETRIS as combination therapy with CHP, treatment emergent neuropathy occurred in 52% of the population; peripheral motor neuropathy occurred in 9% of patients. Peripheral neuropathy led to treatment discontinuation in 1%, dose reductions in 7% and dose delays in <1% of patients. For patients who experienced peripheral neuropathy the median time of onset was 9.1 weeks. Patients who discontinued due to peripheral neuropathy received a median of 5 doses of ADCETRIS+CHP (A+CHP) before discontinuation of one or more agents.
Among patients who experienced peripheral neuropathy, the median follow up time from end of treatment until last evaluation was approximately 177 weeks. At the time of last evaluation, 64% who experienced peripheral neuropathy had resolution or improvement of their peripheral neuropathy symptoms. The median time from onset to resolution or improvement of peripheral neuropathy events was 19.0 weeks (ranged from 0 weeks to 205 weeks).
Infusion-related reactions: Monotherapy IRRs, such as headache, rash, back pain, vomiting, chills, nausea, dyspnoea, pruritus and cough were reported in 13% of patients. Anaphylactic reactions have been reported (see Precautions). Symptoms of an anaphylactic reaction may include, but are not limited to, urticaria, angioedema, hypotension and bronchospasm.
Combination therapy: IRRs, such as headache, rash, back pain, vomiting, chills, nausea, dyspnoea, pruritus, cough, infusion site pain and pyrexia were reported in 8% of patients. Anaphylactic reactions have been reported (see Precautions). Symptoms of an anaphylactic reaction may include, but are not limited to, urticaria, angioedema, hypotension and bronchospasm.
Immunogenicity: In clinical trials, patients were periodically tested for antibodies to brentuximab vedotin using a sensitive electrochemiluminescent immunoassay. There was a higher incidence of infusion-related reactions observed in patients with antibodies to brentuximab vedotin relative to patients who tested transiently positive or negative.
The presence of antibodies to brentuximab vedotin did not correlate with a clinically meaningful reduction in serum brentuximab vedotin levels and did not result in a decrease in the efficacy of brentuximab vedotin. While the presence of antibodies to brentuximab vedotin does not necessarily predict the development of an IRR, there was a higher incidence of IRRs observed in patients with persistently positive anti-drug antibodies (ADA) relative to patients with transiently positive ADA and never positive ADA.
There was a trend of increased clearance of brentuximab vedotin in paediatric patients confirmed positive for ADAs. No patients aged < 12 years (0 of 11) and 2 patients aged ≥ 12 years (2 of 23) became persistently ADA positive.
Paediatric population: Safety was evaluated in a phase 1/2 study in paediatric patients aged 7-17 years of age (n = 36) with relapsed or refractory (r/r) HL and sALCL. In this study in 36 patients, no new safety concerns were reported.
Elderly: Monotherapy: The safety profile in elderly patients is generally in line with that of adult patients. However, elderly patients may be more susceptible to events such as pneumonia, neutropenia and febrile neutropenia.
Combination therapy: In older patients (≥ 60 years of age; n = 186 [21%]), the incidence of adverse events was similar across treatment arms. More serious adverse events and dose modifications (including dose delays, reductions, and discontinuations) were reported in the older patients compared with the overall study population. Advanced age was a risk factor for febrile neutropenia in patients in both arms. Older patients who received G-CSF primary prophylaxis had lower incidence of neutropenia and febrile neutropenia than those who did not receive G-CSF primary prophylaxis.
View ADR Monitoring Form